

GRAPHICAL ABSTRACT
ABSTRACT
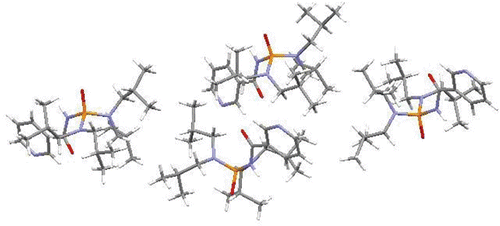
Single crystals of C5H4NC(O)NHP(O)[N(i-C4H9)2]2 were prepared and investigated by X-ray crystallography. Interestingly, four symmetrically independent conformers were detected in the structure of this compound by X-ray crystallography. The greatest difference in these conformers was different torsion angles. In all conformers, the phosphoryl and carbonyl groups showed anti-configurations, and the two terminal CH3 groups of each alkyl chain in amine parts have different spatial orientations due to their connection to a prochiral carbon atom. The diastereotopic protons of every CH2 moiety in amine groups also have different spatial orientations. Every conformer forms a centrosymmetric dimer with its own symmetrically generated analog via a hydrogen bond. The first conformer connects to others (A, B, and C) via electrostatic interactions and forms a tetramer. All hydrogen bonds and electrostatic interactions result in the formation of a three-dimensional polymeric network in the crystalline lattice of compound. To find the most stable conformer, density functional theory (DFT) calculations were performed. The computationally optimized geometric parameters are in good agreement with the experimental results. According to DFT calculations, B is the most stable conformer with energy of −987714.07 kcal/mol. In all conformers the electron density of HOMO is localized on P=O and C=O oxygen atoms and some parts of amine groups, while LUMO is localized on the nicotinamide part of the molecule.
Funding
Support of this work by the Research Councils of the Tarbiat Modares University and the Iranian Research Organization for Science and Technology (IROST) is gratefully acknowledged.