
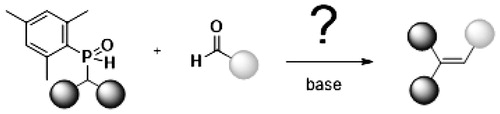
Abstract
Benzhydryl(mesityl)phosphane oxide 2-H is prepared and its deprotonation and nucleophilicity behavior investigated. Treatment of lithium salts of 2-H with MeI results in methylation at the P-center while the benzyl carbon is not affected. Only upon double lithiation, it is possible to methylate also the benzyl carbon. Di-anion 2-2Li is reactive towards benzaldehyde to afford moderate amounts of triphenylethene after basic work.
GRAPHICAL ABSTRACT
Introduction
The importance of C = C double bonds in modern organic chemistry is difficult to overestimate. Alkenes play key roles in polymer and material sciences,[Citation1] are present in biologically active molecules[Citation2] and are essential for pigments.[Citation3] Their synthesis is one of the most fundamental tasks of organic chemistry since the double bond was first postulated by Butlerov in his theory of chemical structure in 1861.[Citation4] Today, there is a large number of methodologies for the preparation of C = C double bonds, for example, by dehydrohalogenation[Citation5] or elimination of water,[Citation6] by rearrangements,[Citation7] or olefination reactions (e.g. Peterson olefination,[Citation8] Wittig reaction,[Citation9] Horner-Wadsworth-Emmons reaction[Citation10]). The approach that is taken in each specific case depends largely on the availability of the starting materials, on the position in the molecule at which the double bond has to be installed, and the reactivity of the remaining part of the molecules. Recently our group published a procedure which allows the formation of C = C doubles bond by the direct coupling of two aldehydes ().[Citation11–13] In contrast to the McMurry protocol[Citation14] which uses low-valent transitional metals, requires harsh reaction conditions, and does not allow the selective preparation of unsymmetric alkenes from the coupling of two non-identical carbonyl compounds, our approach proceeds by a stepwise anionic mechanism which resolves the drawbacks mentioned above. In our synthetic methodology,[Citation11] a first aldehyde reacts with a lithiated phosphanylphosphonate to obtain phosphaalkenes in which the C-center of the P = C double bond exhibits an opposite polarization compared to that in the starting carbonyl compound (). As a consequence of this Umpolung, treatment of the phosphaalkene with a Bu4NOH solution results in the formal addition of water to the P = C double bond with a hydroxyl group placed at the P-center. The thereby obtained hydrophosphinite can be present in solution in two forms[Citation15,Citation16] – either as a trivalent (hydrophosphinic acid) or a pentavalent (secondary phosphine oxide) phosphorus compound. In the presence of a second aldehyde, the secondary phosphine oxide reacts smoothly and an alkene is easily formed within a few minutes. In case a second aldehyde is not present, the crucial secondary phosphine oxide intermediate can be isolated from the reaction mixture. As a proof of the proposed mechanism, it can be subjected in its purified form to the reaction conditions in the presence of the aldehyde to form the olefinic product.
Figure 1. (a) Goal of the project – the reductive coupling of two aldehydes to C = C double bonds, (b) Alkene formation via phosphaalkenes and secondary phosphine oxides, (c) Phosphine oxides used in previous and present work.
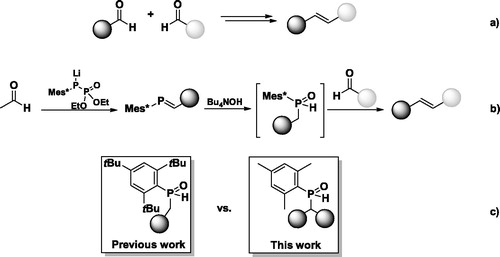
Despite all of the advantages of the developed sequence, several restrictions in substrate scope were observed.[Citation11] Only secondary phosphine oxides obtained from aldehydes bearing electron withdrawing (EWG) substituents appeared to be sufficiently active in the olefination step. The second aldehyde also needs to be selected with care since strongly electron donating aldehydes are not suitable for this reaction. We reasoned that one of the ways to overcome these limitations could be to increase the reactivity of the deprotonated phospine oxides by decreasing the steric bulk around the phosphorus center. Considering all potential synthetic complications that may arise when preparing phosphaalkenes with decreased kinetic stabilization due to the smaller P-substituents,[Citation17] we were interested in scrutinizing the reactivity of benzhydrylphosphane oxides that carry a 2,4,6-trimethylphenyl (mesityl, Mes) P-substituent instead of the previously published 2,4,6-tri(tertbutyl)phenyl (Mes*) group (). As described above, the benzylphosphine oxides are crucial intermediates in the overall coupling sequence, and their downstream chemistry is thus imperative to investigate and understand. We thus set out to prepare and study the deprotonation behavior of benzylmesitylphosphane oxides, to investigate their methylation behavior to identify the most nucleophilic site, and finally check its reactivity towards benzaldehyde.
Results and discussion
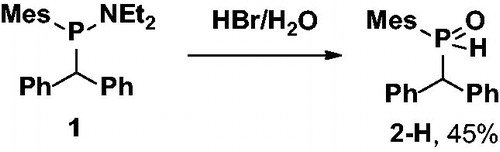
Benzhydryl(mesityl)phosphane oxide 2-H was prepared from literature-known compound 118 by treatment with a concentrated aqueous HBr solution (Scheme 1). The exothermic reaction proceeds smoothly and is completed within several minutes as clearly visible from 31P NMR spectroscopy were the peak of the starting material 1 at +67 ppm completely disappears and a new peak of the product can be found at +24 ppm. Product 2-H is obtained in 45% isolated yield as a white powder.
Scheme 1. Preparation of target secondary phosphine oxide 2-H.
The 1H NMR spectrum of 2-H features a doublet of doublets at 8.10 ppm with coupling constants 1JHP = 477 Hz and 3JHH = 3 Hz. The size of the H-P coupling constant confirms the domination of the pentavalent structure of 2-H in solution. A second doublet of doublets can be found at 4.53 ppm with coupling constants 2JHP = 14 Hz and 3JHH = 3 Hz that is assigned to the proton at the α benzyl carbon.
Having 2-H in hand, a series of experiments were conducted to reveal the compound’s primary deprotonation site. Secondary phosphine oxide 2-H was thus treated with one or two equivalents of BuLi in THF (Scheme 2).
Scheme 2. Reaction of 2-H with BuLi and CH3I.
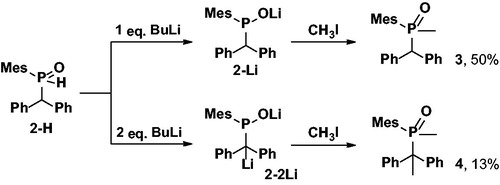
Addition of 1 eq. of base resulted in almost no color change of the reaction mixture and the 31P NMR spectrum indicated the formation of a new species with a chemical shift of +104 ppm which is consistent with the lithium salt of the hydrophosphinic acid 2-Li (resonance structure B in ). Treatment of 2-Li with excess methyl iodide results in the formation of a single product according to the 31P NMR spectroscopy with a chemical shift of +46 ppm (THF solution). In the 1H NMR spectrum of the isolated product, the large doublet of doublets at 8.10 ppm is not observed any longer, indicating the absence of a proton that is directly attached to the phosphorus center, suggesting the formation of the tertiary phosphine oxide 3. The proton at the α carbon appears as a doublet at 4.42 ppm with 2JHP = 9 Hz indicating that C-centered alkylation has not occured. A new doublet at 1.74 ppm with 2JHP = 12 Hz and integrating for 3 protons can be assigned to the methyl group introduced at the phosphorus center, suggesting reactivity through intermediate A (). All together, the NMR spectroscopic data suggests exclusive alkylation at the P-center by a Michaelis - Becker reaction and, indeed, compound 3 appears to be literature known with analytical data fully consistent with ours.[Citation19] Treatment of 2-Li with aldehydes resulted in no reaction and starting materials were isolated even under prolonged reaction times and elevated temperatures. This result indicates the complete absence of anions that could be described by resonance structures C and D in solutions of 2-Li ().
Figure 2. Possible structures of anions present in reaction mixtures after mono and di-lithiation of 2-H.
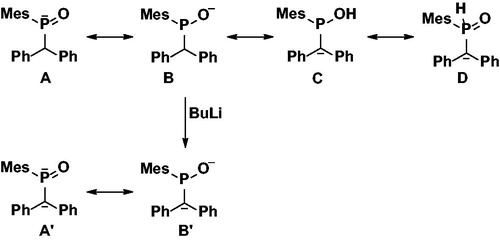
Treatment of 2-H with two equivalents of BuLi resulted in a strong color change from colorless to bright orange and finally red solutions. The reaction mixture showed multiple very broaden signals at ca. +105 ppm in the 31P NMR spectra, indicating complex and fast equilibria at room temperature (, resonance structures A’ and B’). Treatment of 2-2Li with methyl iodide resulted in the formation of product 4 (Scheme 2, δ = +58 ppm) as the major product with small trace of 3 also visible in the 31P NMR spectrum. The 1H NMR spectrum of 4 contains no signals in the region from 4 to 6 ppm which could be assigned to the proton at the α carbon, but features instead two doublets at 1.97 ppm (2JHP = 16 Hz) and 1.62 ppm (3JHP = 7 Hz) from the methyl groups at the P and C-centers, respectively.
Encouraged by the results described above, we decided to treat solution of 2-2Li with benzaldehyde (Scheme 3).
Scheme 3. Reaction of 2-2Li with benzaldehyde affording 5 in 11% yield.
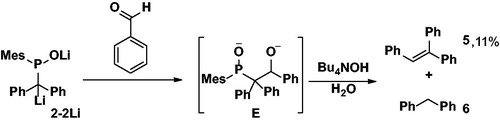
After several hours at room temperature the color of the reaction mixture changed from bright orange-red to pale yellow, indicating complete consumption of 2-2Li. Unfortunately, no triphenylethene could be identified in the reaction mixture. We hypothesize that intermediate anion E (Scheme 3) should have been formed, but due to the negative charge at the P-center, it cannot undergo cyclization to the oxaphosphetane intermediate that is crucial for alkene formation. This problem could be overcome by addition of an aqueous solution of Bu4NOH to the reaction mixture to provide a pH at which the P–O− is presumably protonated to allow the alcohol at the β carbon to act as a nucleophile at the P-center. Thereby, the oxaphosphetane is formed which finally collapses to form the desired product 5, as evidenced by analytical data that are fully consistent with the literature values.[Citation20] Unfortunately, compound 5 was only isolated in 11% yield, with diphenylmethane 6 as the main byproduct (Scheme 3). The dianionic character of 2-2Li presumably aids in its decomposition with the diphenylmethyl anion being a good leaving group that is then protonated upon aqueous work-up to form 6.
Conclusions
A method for the easy preparation of 2-H from literature known compound was developed. Secondary phosphine oxide 2-H appears to undergo stepwise deprotonation upon addition of one or two equivalents of base. First deprotonation is shown to occur at the phosphorus center and the obtained anion reacts selectively to give the Michaelis – Becker product 3. The second deprotonation takes place at the α carbon and the obtained dianion 2-2Li can be successfully double methylated. Treatment of 2-2Li with benzaldehyde gives rise to the desired triphenylethene, albeit only in 11% isolated yield after basic work up. Overall, 2-H shows very different reactivity in comparison to its Mes* analog and even though its double lithium salt 2-2Li can be used for coupling reactions to obtain the alkenes, the current procedure is not optimal and alternative approaches to increase the scope of the aldehyde-aldehyde coupling needs to be found. These may include future work to decrease the steric demand of the P-substituent further, possibly together with an increase of oxygenation level of the P-center.[Citation12,Citation13]
Experimental
General experimental procedures. 1H, 13C and 31P NMR spectra were recorded on a 400 MHz spectrometer (JEOL) unless noted otherwise. Chemical shifts were referenced to residual solvent peaks and are given as follows: chemical shift (δ, ppm), multiplicity [s, singlet; br, broad; d, doublet, t, triplet, q, quartet, m, multiplet, coupling constant (Hz), integration]. All compounds displayed the expected isotope distribution patterns. Anhydrous CH2Cl2 was obtained by distillation from CaH2 under an N2 atmosphere. Anhydrous THF and Et2O were distilled from sodium under an N2 atmosphere. Compound 1 was prepared according to literature procedures.[Citation18]
Benzhydryl(mesityl)phosphane oxide 2-H
To a cooled (ca 4 °C) solution of 4 g (0.01 mol) of 1 in 50 mL DCM, concentrated solution of HBr in water (45%, 0.03 mol, 1.25 eq., 1.5 mL) was added dropwise. The reaction mixture was stirred for 30 min at r.t., diluted with DCM (total volume ca 150 mL) and washed with water until the pH of the organic phase was found neutral. The organic phase was dried over MgSO4, filtered and the solvent removed under vacuum to afford crude 2-H as a white solid. Washing of this solid with 2 × 25 mL Et2O and drying under vacuum gave the final product as a white fine powder. Yield 45%, 1.5 g. 1H NMR (400 MHz, CDCl3): δ = 8.10 (dd, 1JHP = 477, 3JHH = 3 Hz, 1H, PH), 7.38–7.33 (m, 4H, Ph), 7.33–7.22 (m, 6H, Ph), 6.78 (d, 4JHP = 4 Hz, 2H, Mes), 4.53 (dd, 2JHP = 14, 3JHH = 3 Hz, 1H, PCH), 2.27 (s, 3H, p-Me-Mes), 2.17 (s, 6H, o-Me-Mes) ppm. 31P NMR (162 MHz, CDCl3): δ = 25.2 ppm. 13C NMR (101 MHz, CDCl3): δ = 142.4 (s), 142.4 (s), 142.2 (d, J = 10 Hz), 137.1 (d, J = 3 Hz), 135.7 (d, J = 6 Hz), 130.3 (d, J = 11 Hz), 130.0 (s), 129. 9 (s), 129.5 (s), 129.4 (m), 128.9 (d, J = 2 Hz), 128.8 (d, J = 1 Hz), 122.8 (d, J = 97 Hz), 54.2 (d, J = 61 Hz), 21.3 (d, J = 1 Hz), 21.3 (s), 21.2 (s) ppm. HR-MS/ESI (+): m/z = 335.1657 [M + H]+, calculated [C22H24OP]+ = 335.1565.
Benzhydryl(mesityl)(methyl)phosphane oxide 3
To a cooled (ca −40 °C) solution of 50 mg (0.15 mmol) of 2-H in 5 mL THF, a hexane solution of n-BuLi (1.6 M, 0.16 mol, 1.1 eq.) was added dropwise. The pale yellow reaction mixture was stirred for 10 min at −40 °C and then it was stirred at ambient temperature for an additional hour. Excess of MeI was added to the reaction mixture at ambient temperature and an aliquote of the mixture was analyzed by 31P NMR with an internal benzene-d6 capillary. A new peak of the product 3 (δ = + 46 ppm) is observed. The reaction is quenched with water and the aqueous phase was extracted with DCM (×3). The organic phase was dried over MgSO4, filtered and the solvent removed under vacuum to afford crude 3 as a white solid. Product 3 was isolated via silica gel column chromatography (DCM:acetone =18:1, Rf = 0.37) as a white solid. Yield 50%, 26 mg. 1H NMR (400 MHz, CDCl3): δ = 7.77–7.72 (m, 2H, Ph), 7.41–7.35 (m, 2H, Ph), 7.34–7.26 (m, 2H, Ph), 7.18–7.14 (m, 2H, Ph), 7.13.10 (m, 2H, Ph), 6.79 (d, 4JHP = 3.4 Hz, 2H, Mes), 4.42 (d, 2JHP = 8.4 Hz, 1H, PCH), 2.34 (s, 6H, o-Me-Mes), 2.24 (s, 3H, p-Me-Mes), 1.74 (d, 2JHP = 12.2 Hz, 3H, PCH3). 31P NMR (162 MHz, CDCl3): δ = 42.7 ppm. 13C NMR (101 MHz, CDCl3): δ = 141.3 (s), 141.3 (s), 137.5 (d, J = 4 Hz), 136.5 (d, J = 6 Hz), 131.2 (d, J = 11 Hz), 130.1 (s), 130.1 (s), 129.4 (d, J = 5 Hz), 128.9 (s, J = 2 Hz), 128.2 (d, J = 2 Hz), 127.5 (s), 127.5 (s), 126.9 (d, J = 3 Hz), 125.8 (d, J = 92 Hz), 55.9 (d, J = 61 Hz), 23.6 (d, J = 3 Hz), 21.0 (d, J = 1 Hz), 20.1 (d, J = 68 Hz) ppm. HR-MS/ESI (+): m/z = 349.1887 [M + H]+, calculated [C23H26OP]+ = 349.1721.
(1,1-diphenylethyl)(mesityl)(methyl)phosphane oxide 4
To a cooled (ca −40 °C) solution of 30 mg (0.089 mmol) of 2-H in 5 mL THF, a hexane solution of n-BuLi (1.6 M, 0.19 mmol, 2.2 eq.) was added dropwise. The deep orange reaction mixture was stirred for 30 min at -40 °C and then it was stirred at ambient temperature for additional 2.5 hours. Excess of MeI was added to the reaction mixture at ambient temperature and an aliquote of the mixture was analyzed by 31P NMR with an internal benzene-d6 capillary. A new peak of the product 4 (δ = + 58 ppm) is detected, with the presence also of some 3 (Ratio 4: 3 obtained by 31P NMR is 1: 0.2). The solvent removed under vacuum to afford crude 4 as a white solid. NMR and HRMS measurements were performed directly on the crude mixture, without any further purification. Crude yield of 4 estimated 13%. 1H NMR (400 MHz, CDCl3): δ = 7.45–7.13 (m, 10 H, Ph), 6.75 (d, 4JHP = 3.4 Hz, 2H, Mes), 2.24 (s, 6H, o-Me-Mes), 2.19 (s, 3H, p-Me-Mes), 1.97 (d, 2JHP = 15.8 Hz, 3H, PCH3), 1.62 (d, 3JHP = 7.2 Hz, 3 H, PCCH3) ppm. 31P NMR (162 MHz, CDCl3): δ = 58.4 ppm. HR-MS/ESI (+): m/z = 363.2031 [M + H]+, calculated [C24H28OP]+ = 363.1878.
Triphenylethene 5
To a cooled (ca −40 °C) solution of 100 mg (0.3 mmol) of 2-H in 15 mL THF, a hexane solution of n-BuLi (2.5 M, 0.6 mmol, 2 eq.) was added dropwise. The deep orange reaction mixture was stirred for 30 min at −40 °C and then it was stirred at ambient temperature for additional 2.5 hours. 34 mg (0.32 mmol, 1.05 eq) of benzaldehyde were added to the reaction mixture that was stirred at ambient temperature for additional 12 hours. Excess of an aqueous solution of TBAOH was added. The aqueous phase was extracted with EtOAc (×3). The organic phase was dried over MgSO4, filtered and the solvent removed under vacuum to afford a crude mixture of 5, 6 (relative ratio 5:6 = 1:2) and other undefined byproducts. The crude was subjected to a silica gel column chromatography (10% of Et2O in heptane, Rf = 0.7) to afford a mixture of 5 and 6 in 2: 1 ratio. Yield of 5 11%, 8 mg. 1H NMR of 5 and 6 are in accordance with literature[Citation20] and commercial available product, respectively.
Acknowledgments
Financial support for this work from the Swedish Research Council is gratefully acknowledged.
References
- Marder, S. R.; Beratan, D. N.; Cheng, L. T. Approaches for Optimizing the First Electronic Hyperpolarizability of Conjugated Organic Molecules. Science 1991, 252, 103–106.
- De Filippis, B.; Ammazzalorso, A.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Amoroso, R. Anticancer Activity of Stilbene-based Derivatives. Chem. Med. Chem. 2017, 12, 558–570.
- Manfredi, N.; Cecconi, B.; Abbotto, A. Multi-Branched Multi-Anchoring Metal-Free Dyes for Dye-Sensitized Solar Cells. Eur. J. Org. Chem. 2014, 2014, 7069–7086.
- Larder, D. F.; Kluge, F. F. Alexander Mikhailovich Butlerov's Theory of Chemical Structure. J. Chem. Educ. 1971, 48, 287–289.
- Bissember, A. C.; Levina, A.; Fu, G. C. A Mild, Palladium-Catalyzed Method for the Dehydrohalogenation of Alkyl Bromides: Synthetic and Mechanistic Studies. J. Am. Chem. Soc. 2012, 134, 14232–14237.
- Friesen, J. B.; Schretzman, R. Dehydration of 2-Methyl-1-Cyclohexanol: New Findings from a Popular Undergraduate Laboratory Experiment. J. Chem. Educ. 2011, 88, 1141–1147.
- Enders, D.; Knopp, M.; Schiffers, R. Asymmetric [3.3]-Sigmatropic Rearrangements in Organic Synthesis. Tetrahedron: Asymmetry 1996, 7, 1847–1882.
- Staden, L. F. v.; Gravestock, D.; Ager, D. New Developments in the Peterson Olefination Reaction. Chem. Soc. Rev. 2002, 31, 195–200.
- Wittig, G. From Diyls to Ylides to my Idyll. Science 1980, 210, 600–604.
- Boutagy, J.; Thomas, R. Olefin Synthesis with Organic Phosphonate Carbanions. Chem. Rev. 1974, 74, 87–99.
- Esfandiarfard, K.; Mai, J.; Ott, S. Unsymmetrical E-Alkenes from the Stereoselective Reductive Coupling of Two Aldehydes. J. Am. Chem. Soc. 2017, 139, 2940–2943.
- Mai, J.; Arkhypchuk, A. I.; Gupta, A. K.; Ott, S. Reductive Coupling of Two Aldehydes to Unsymmetrical E-Alkenes via Phosphaalkene and Phosphinate Intermediates. Chem. Commun. (Camb.) 2018, 54, 7163–7166.
- Arkhypchuk, A. I.; D'Imperio, N.; Ott, S. One-Pot Intermolecular Reductive Cross-Coupling of Deactivated Aldehydes to Unsymmetrically 1,2-Disubstituted Alkenes. Org. Lett. 2018, 20, 5086–5089.
- McMurry, J. E. Carbonyl-Coupling Reactions Using Low-Valent Titanium. Chem. Rev. 1989, 89, 1513–1524.
- Christiansen, A.; Li, C.; Garland, M.; Selent, D.; Ludwig, R.; Spannenberg, A.; Baumann, W.; Franke, R.; Börner, A. On the Tautomerism of Secondary Phosphane Oxides. Eur. J. Org. Chem. 2010, 2010, 2733–2741.
- Kurscheid, B.; Wiebe, W.; Neumann, B.; Stammler, H. G.; Hoge, B. Investigations of the Tautomeric Equilibria between Phosphane Oxides and Their Corresponding Phosphinous Acids Bearing Electron-Withdrawing Perfluoroaryl Groups. Eur. J. Inorg. Chem. 2011, 2011, 5523–5529.
- Wang, S.; Samedov, K.; Serin, S. C.; Gates, D. P. PhP = CPh 2 and Related Phosphaalkenes: A Solution Equilibrium Between a Phosphaalkene and a 1,2-Diphosphetane. Eur. J. Inorg. Chem. 2016, 2016, 4144–4151.
- Van Der Knaap, T. A.; Klebach, T. C.; Visser, F.; Bickelhaupt, F.; Ros, P.; Baerends, E. J.; Stam, C. H.; Konijn, M. Synthesis and Structure of Aryl-Substituted Phospha-Alkenes. M. Tetrahedron 1984, 40, 765–776.
- Van der Knap, T. A.; Bickelhaupt, F. A Nucleophilic Reaction of a Phosphaalkene: The Methylation of Mesityldiphenylmethylenephosphine. Tetrahedron Lett. 1982, 23, 2037–2040.
- Bi Jie, L.; Yi Zhou, L.; Xing Yu, L.; Jia, L.; Bing Tao, G.; Zhang Jie, S. Cross-Coupling of Aryl/Alkenyl Pivalates with Organozinc Reagents through Nickel-Catalyzed C-O Bond Activation under Mild Reaction Conditions. Angew. Chem. Int. Ed. Engl. 2008, 47, 10124–10127.