
Abstract
Parsaclisib, a potent, highly selective, next-generation PI3Kδ inhibitor, was evaluated as monotherapy in CITADEL-202 (NCT02998476), an open-label, multicenter, phase 2 study in patients with relapsed or refractory diffuse large B-cell lymphoma. Patients enrolled into 2 groups (A, Bruton tyrosine kinase [BTK] inhibitor naïve, n = 55; B, BTK inhibitor experienced, n = 5) received oral parsaclisib 20 mg once daily for 8 weeks, then 20 mg once weekly while deriving benefit. The futility boundary was crossed at the interim analysis of Group A, resulting in a negative study. Parsaclisib monotherapy demonstrated an objective response rate (ORR) of 25.5% (8 complete metabolic responses/6 partial metabolic responses) and a median duration of response of 6.2 months. ORR in Group B was 20.0% (1 complete metabolic response). Parsaclisib monotherapy demonstrated manageable toxicities with no new safety signals reported. Further evaluation of parsaclisib in combination with standard therapies and active investigational agents is underway.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is an aggressive form of non-Hodgkin lymphoma (NHL), accounting for approximately 30–40% of all NHL cases [Citation1]. Distinct molecular forms of DLBCL include activated B-cell–like (ABC) and germinal center B-cell–like (GCB) [Citation2]. The current standard first-line treatment for patients with previously untreated DLBCL is immunochemotherapy with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) [Citation3]; however, 15–30% of patients relapse or remain refractory to initial immunochemotherapy [Citation4]. For eligible patients with relapsed or refractory DLBCL, second-line therapy typically includes salvage chemotherapy followed by high-dose chemotherapy with autologous stem cell transplant (SCT) [Citation3–5]. Ibrutinib, a Bruton tyrosine kinase (BTK) inhibitor, is not US Food and Drug Administration–approved for DLBCL but is recommended in the National Comprehensive Cancer Network guidelines as a second-line treatment option for patients with non-GCB DLBCL [Citation3,Citation6]. Chimeric antigen receptor T-cell therapy or polatuzumab vedotin plus bendamustine plus rituximab are additional treatment options for patients with relapsed or refractory DLBCL, with specific limitations [Citation3,Citation7]. Prognosis is poor in patients whose disease fails to respond to both first-line and second-line therapies (median overall survival [OS], 3.6 and 4.4 months, respectively) [Citation8]. Therefore, effective treatment options are needed for patients with relapsed or refractory DLBCL [Citation4].
The B-cell receptor signaling pathway is a key driver of pathogenesis in human B-cell malignancies [Citation9,Citation10]. Constitutive signaling through B-cell receptors leads to activation of Class I phosphoinositide 3-kinase (PI3K) and other downstream signaling pathways [Citation10,Citation11]. The Class I PI3K family includes the catalytic p110 α, β, γ, and δ isoforms [Citation12]. Aberrant activation of the PI3Kδ isoform is associated with cellular activation, proliferation, and survival in B-cell malignancies [Citation12,Citation13]. Clinical studies in relapsed and/or refractory NHL subtypes have led to regulatory approval for the PI3K inhibitors idelalisib (relapsed follicular lymphoma [FL], chronic lymphocytic leukemia, or small lymphocytic lymphoma) [Citation14], duvelisib (relapsed or refractory FL, chronic lymphocytic leukemia, or small lymphocytic lymphoma) [Citation15], and copanlisib (relapsed FL) [Citation16]. However, clinical utility of the first-generation PI3K inhibitors (e.g. idelalisib and duvelisib) has been limited owing to their safety profiles [Citation14,Citation15].
Parsaclisib (INCB050465) is a potent and highly selective next-generation PI3Kδ inhibitor with approximately 20,000-fold selectivity for PI3Kδ over other isoforms PI3Kα, PI3Kβ, and PI3Kγ. It has a whole blood half-maximal inhibitory concentration of 10 nM, and a 90% maximal inhibitory concentration of 77 nM [Citation17–19]. In a phase 1/2 study of parsaclisib in heavily pretreated patients with relapsed or refractory NHL, parsaclisib monotherapy demonstrated promising preliminary antitumor activity in patients with DLBCL, with an objective response rate (ORR) of 30.4% and a median duration of response (DOR) of 13.5 months. No unexpected safety findings were observed [Citation19]. The purpose of the present study was to assess further the efficacy and safety of parsaclisib monotherapy in patients with relapsed or refractory DLBCL.
Methods
Study design
This open-label, phase 2, multicenter, international study (CITADEL-202; ClinicalTrials.gov identifier: NCT02998476) enrolled patients with relapsed or refractory DLBCL into 2 groups based on whether patients received or did not receive prior treatment with a BTK inhibitor (e.g. ibrutinib). Group A included patients who were not previously treated with a BTK inhibitor and Group B included patients who were previously treated with a BTK inhibitor. Clinical outcomes after treatment with ibrutinib in DLBCL are not fully known; therefore, cohorts were separated based on prior treatment with a BTK inhibitor in case BTK-inhibitor resistance mechanisms could not be overcome by a PI3Kδ inhibitor.
The study was approved by each site’s ethics review board and conducted in accordance with the International Council for Harmonisation guideline for Good Clinical Practice, the Declaration of Helsinki, and applicable local ethical and legal requirements. All patients provided written, informed consent before study participation.
Parsaclisib was administered orally at 20 mg once daily (QD) for 8 weeks followed by 20 mg once weekly (QW). Treatment continued until disease progression, unacceptable toxicity, withdrawal of consent, or death (Supplementary Figure 1). A protocol amendment (23 February 2017) required all patients to receive an investigator-determined standard Pneumocystis jirovecii pneumonia (PJP) prophylaxis regimen during study treatment for 2–6 months after the last dose of study treatment.
Study population
Eligible patients were ≥18 years of age (≥19 years in South Korea); had histologically documented relapsed or refractory DLBCL (defined as having received ≥2 but no more than 5 prior regimens [initial study protocol allowed patients receiving ≥1 prior regimen to be enrolled] and were ineligible for high-dose chemotherapy/autologous SCT); had ≥1 measurable nodal lesion (≥2 cm in longest dimension) or ≥1 measurable extranodal lesion (>1 cm in longest dimension) by computed tomography (CT) scan or magnetic resonance imaging (MRI); had an Eastern Cooperative Oncology Group performance status ≤2; were willing to undergo lymph node biopsy of accessible adenopathy, or had provisions of most recent, available archive tumor biopsy (for potential biomarker analysis); had adequate hematologic, hepatic, and renal function; and had received prior BTK inhibitor therapy (Group B only). Cell of origin analysis was performed locally to identify GCB and ABC/non-GCB DLBCL subtypes.
Exclusion criteria included primary mediastinal large B-cell lymphoma or known brain/central nervous system metastases; history of uncontrolled seizures; allogeneic SCT within ≤6 months, active graft versus host disease post-allogeneic transplant, or autologous SCT within ≤3 months before the first dose of study drug; New York Heart Association Class II–IV congestive heart failure or uncontrolled arrhythmia; known human immunodeficiency virus infection or positive immunoassay; immunosuppressive therapy ≤28 days of the date of study drug administration; concurrent anticancer therapy such as biologic, hormonal, investigational, chemotherapy, radiotherapy, immunotherapy, surgery, or tumor embolization. Use or expected use of potent cytochrome P450 3A4 inhibitors or inducers within 14 days or 5 half-lives (whichever is longer), before study drug commencement was prohibited. Prior treatment with PI3Kδ-selective or pan-PI3K inhibitors was excluded for both Groups A and B.
Study outcomes and assessments
The primary endpoint was ORR in Group A, defined as the percentage of patients with a complete metabolic response (CMR) or partial metabolic response (PMR). Response was assessed by an independent review committee (IRC) using the positron emission tomography (PET)-CT response criteria of the Lugano Classification [Citation20], and evaluated every 9 (±1) weeks through week 27, and every 18 (±1) weeks thereafter until disease progression, commencement of a new anticancer therapy, or death, whichever occurred first. Secondary endpoints included DOR, progression-free survival (PFS), and OS in Group A, and safety in Groups A and B. DOR and PFS were determined by radiographic disease assessments provided by an IRC. Safety was assessed via vital signs, physical examination, 12-lead electrocardiograms, chemistry and hematology laboratory values, and adverse events (AEs). AEs were tabulated by Medical Dictionary for Regulatory Activities preferred term and system organ class, and severity assessed using National Cancer Institute Common Technology Criteria for Adverse Events version 4.03.
Exploratory endpoints included parsaclisib population pharmacokinetics (PK) (apparent clearance following oral dose administration [CL/F] and volume of distribution). A preliminary population PK model was developed using parsaclisib plasma concentrations measured at protocol-specified time points from this trial (CITADEL-202) and the CITADEL-101 trial (NCT02018861; conducted in patients with relapsed or refractory B-cell malignancies, including DLBCL). The population PK model consisted of a 2-compartment disposition model with first-order absorption and first-order elimination. PK plasma samples were obtained on study visit day 1 (predose, 30 [±10] minutes, and 120 [±10] minutes) and day 15 (predose, 60 [±10] minutes, and 4 [±0.5] hours). Parsaclisib concentration in plasma samples was measured by Incyte Corporation (Wilmington, DE, USA) using a validated liquid chromatography–tandem mass spectrometry assay.
Statistical analysis
Efficacy and safety were reported for patients who received ≥1 dose of parsaclisib. PK were assessed for patients who received ≥1 dose of parsaclisib and provided ≥1 postdose plasma sample. ORR was estimated with 95% confidence intervals (CIs) calculated using the exact method for binomial distributions. The Kaplan–Meier estimations of median DOR, PFS, and OS with their 95% CIs were determined.
Planned enrollment in Group A was 100 patients. Assuming the true ORR is 50%, then there is an approximately 90% probability of observing the lower bound of the 95% CI for ORR to be ≥35%. For Group B, up to 20 patients were to be enrolled, with an 87% probability of observing ≥6 responders if the true ORR in the group is 40%. An interim analysis was planned when the first 40 patients in Group A had been treated and evaluated for response or had permanently discontinued study treatment owing to disease progression, consent withdrawal, or death; if ≤13 (≤32.5%) of the 40 patients responded (i.e. CMR or PMR) by an IRC assessment, enrollment into Group A of the study was to be terminated for futility. No formal interim analysis was planned for Group B.
Results
Patient demographics and disease characteristics
Between 2 March 2017 and 10 April 2018, 60 patients from 33 study centers in 11 countries were treated. As of the 22 February 2019 data cutoff, 55 patients were treated in Group A and 5 patients were treated in Group B. Patient demographics and disease characteristics at baseline are presented in . The median (range) age was 71 (36–94) years in Group A and 69 (50–86) years in Group B. In both groups, most patients had 3–5 prior systemic therapies, and most patients had de novo DLBCL. The prior histologies of the patients with transformed disease in Group A (n = 16) were follicular lymphoma (n = 10), marginal zone lymphoma (n = 3), and high grade (large cell) B-cell NHL, NHL B phenotype centrocyte-like diagnosis, and small lymphocytic lymphoma (n = 1 each).
Table 1. Patient demographics and disease characteristics at baselinea.
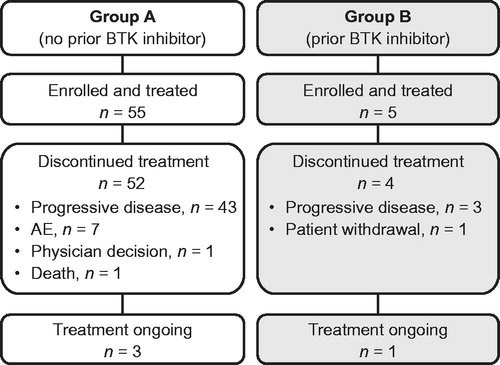
In Group A, the proportion of patients with GCB and ABC/non-GCB subtypes was 40.0% (n = 22) and 34.5% (n = 19), respectively; in Group B, the corresponding proportions were 20.0% (n = 1) and 80.0% (n = 4), respectively (). Fifty-two (94.5%) and 4 (80.0%) patients in Groups A and B, respectively, discontinued study treatment, primarily owing to disease progression (Group A: n = 43 [78.2%]; Group B: n = 3 [60.0%]) (). The median (range) duration of treatment was 1.9 months (0.5–16.0) in Group A and 1.9 months (0.4–12.3) in Group B.
Figure 1. Patient disposition. AE: adverse effect; BTK: Bruton tyrosine kinase.
Efficacy
Planned interim analysis
At the planned interim efficacy analysis in Group A (n = 40), the ORR (by PET) was 25.0% (10 of 40 patients; 5 patients had CMR and 5 patients had PMR) (). The median PFS was 2.1 months (95% CI: 1.9–4.1). As the futility boundary was crossed based on results of the interim analysis, patient recruitment was stopped in both groups.
Table 2. Summary of overall response by IRC-PETa (Group Ab).
Group A
At the data cutoff date, objective response was observed in 14 of 55 patients (25.5%; 95% CI: 14.7–39.0) based on radiologic assessments provided by the IRC using PET, with 10 of the 14 responders (71.4%) achieving a response at the first disease assessment. The median time to response was 9.1 weeks. An objective response was observed in 4 of 22 patients (18.2%) with GCB, 6 of 19 (31.6%) with ABC/non-GCB, and 4 of 14 (28.6%) with unknown subtype. The ORR was 31.3% (95% CI: 11.0–58.7) in patients with transformed disease.
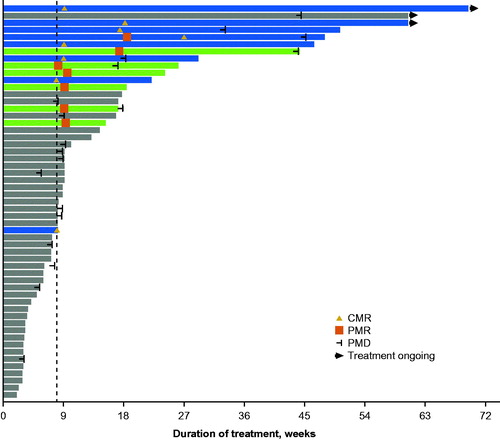
Thirteen patients (23.6%) achieved >50% reduction in target lesion size from baseline as determined by the IRC using CT/MRI (). Based on assessments provided by the IRC using PET, the median PFS was 2.2 months (95% CI: 2.0–4.1), the median OS was 7.0 months (95% CI: 3.5–not reached), and the median DOR was 6.2 months (95% CI: 2.1–not reached). Five patients demonstrated durable responses (>6 months) and 2 responders had treatment ongoing at data cutoff; the longest time on study treatment observed at the time of data cutoff was 16.0 months ().
Figure 2. Best percentage change from baseline in target lesion size (Group A). Percentage change shown for patients with postbaseline assessments from IRC by CT/MRI. CT: computed tomography; IRC: independent review committee; MRI: magnetic resonance imaging. aIndicates patients with best percentage change >100%.
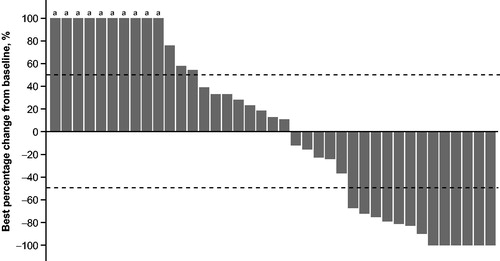
Figure 3. Duration of treatment (Group A). Blue bars indicate patients who achieved CMR and green bars indicate patients who achieved PMR as best overall responses by IRC-PET. Dashed line indicates scheduled switch from once-daily to once-weekly dosing per protocol. CMR: complete metabolic response; IRC: independent review committee; PET: positron emission tomography; PMD: progressive metabolic disease; PMR: partial metabolic response.
When assessed by disease status based on radiologic assessments provided by the IRC using PET, an objective response was observed in 8 of 10 (80.0%; 95% CI: 44.4–97.5) patients with relapsed disease (5 CMR; 3 PMR), 5 of 41 (12.2%; 95% CI: 4.1–26.2) patients with refractory disease (2 CMR; 3 PMR), and 1 of 4 (25.0%; 95% CI: 0.6–80.6) patients with unknown relapsed/refractory disease status (1 CMR).
Group B
Enrollment in Group B was terminated after the interim efficacy analysis in Group A, as a higher ORR was not anticipated in patients with prior BTK inhibitor exposure. Based on the assessments of the 5 patients in Group B provided by the IRC at data cutoff date, 1 patient (non-GCB) achieved CMR (ORR, 20.0%), 3 patients (non-GCB) had progressive disease (assessed by PET in 1 patient and CT/MRI in 2 patients), and 1 patient (GCB) had no postbaseline assessment.
Safety
All treated patients in Groups A and B comprised the safety population (n = 60). Nonhematologic treatment-emergent adverse events (TEAEs) occurring in ≥10% of the patients were primarily grade 1/2 (); rash (any grade: 13 [21.7%]; grade 3/4: 1 [1.7%]) and colitis/diarrhea (any grade: 12 [20.0%]; grade 3/4: 4 [6.7%]) events were the most common nonhematologic TEAEs, reported in ≥20% of patients. Hyperglycemia was reported by 3 patients (5.0%) in the study, with grade 3 hyperglycemia reported in 1 patient (1.7%); no grade 4 hyperglycemia was reported. Hypertension was reported by 1 patient (1.7%; grade 1) in the study, no grade 3/4 hypertension was reported. Severe (grade 3/4) events of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) elevations occurred in 3 (5.0%) and 2 (3.3%) patients, respectively. New or worsening grade 3/4 hematologic TEAEs of neutropenia, anemia, and thrombocytopenia were reported in 4 (6.7%), 3 (5.0%), and 2 (3.3%) patients, respectively (). Serious TEAEs were reported in 41 patients (68.3%), the most frequent (>5%) serious TEAEs were abdominal pain, hypercalcemia, and pyrexia (6.7% each). Enterocolitis (grade 3) and PJP (grade 3) occurred in 1 patient, and colitis (grade 3) occurred in 1 patient.
Table 3. Nonhematologic TEAEs and new or worsening hematologic abnormalities occurring in ≥10% of patients (safety population).
TEAEs led to dose interruption in 24 patients (40.0%; treatment-related in 12 patients), dose reduction in 4 patients (6.7%; all treatment-related; neutropenia [n = 1], maculopapular rash [n = 1], maculopapular rash plus pruritic rash [n = 1], pyrexia plus colitis [n = 1]), and treatment discontinuation in 8 patients (13.3%; treatment-related in 2 patients; ALT and AST increase [n = 1], macular rash [n = 1]). TEAEs leading to death occurred in 7 patients (11.7%; general physical health deterioration [n = 2], pleural effusion, acute respiratory failure, cholestatic jaundice, disease progression, multiple organ dysfunction syndrome [each n = 1]), none was deemed related to parsaclisib.
The parsaclisib QW dosing regimen was administered to 28/55 (51%) patients in Group A; median (range) duration of exposure for QW dosing regimen was 9.0 (1–62) weeks. Grade ≥3 TEAEs occurred in 17/28 patients (60.7%) in Group A during QW dosing period; the most frequently reported Grade ≥3 TEAEs were neutrophil count decreased (10.7%), abdominal pain, general physical health deterioration, pyrexia, increased ALT, increased AST, and increased blood bilirubin in 2 patients (7.1%) each. During QW dosing, dose interruptions were reported in 7/28 (25.0%) of patients in Group A; the most frequently reported reasons were diarrhea and maculopapular rash (7.1% each), pyrexia, CMV infection, AST increased, neutrophil count decreased, exfoliative dermatitis, pruritic rash, and hypotension (3.6% each).
Parsaclisib population pharmacokinetics
Population PK assessments based on this study and CITADEL-101 demonstrated a typical CL/F of 3.11 L/h (for patients with body weight of 79.8 kg) (Supplementary Table 1). PK results also indicated that body weight significantly predicted CL/F and central volume of distribution. No clinically meaningful effect on parsaclisib exposure was observed based on sex, age, body weight, renal function, or liver function.
Discussion
In the phase 1/2 CITADEL-101 study, parsaclisib monotherapy demonstrated an ORR of 30.4%, with a median DOR of 13.5 months (95% CI: 8.3–18.8) in 23 patients with relapsed or refractory DLBCL [Citation19]. Based on these results, we further evaluated the efficacy of parsaclisib monotherapy in patients with relapsed or refractory DLBCL with or without prior BTK inhibitor treatment in this phase 2 study.
At the interim analysis of Group A, the futility boundary was crossed resulting in a negative study. The ORR of 25.5%, which includes 8 CMRs (14.5%), and a median DOR of 6.2 months demonstrates durable monotherapy activity of parsaclisib in relapsed or refractory DLBCL. Notably, 1 patient had a DOR of more than 12.5 months and 3 patients were still ongoing with study treatment at the time of the data cutoff (22 February 2019; ). These response rates with parsaclisib in CITADEL-202 may, in part, be due to the heavily pretreated nature of the patients enrolled. A similar trend was observed in SCHOLAR-1, an international, retrospective patient-level pooled analysis of response rates and survival outcomes of salvage chemotherapy in 636 patients with heavily pretreated refractory DLBCL (defined as no response >4 cycles of first-line therapy or 2 cycles of later-line therapy or relapse ≤12 months after autologous SCT). The study reported a pooled ORR of 26% (pooled complete response, 7%) with a median OS of 6.3 months [Citation21]. In the current study, although Group A and Group B (previously treated with a BTK inhibitor: e.g. ibrutinib) were opened for enrollment at the same time, only 5 patients were enrolled in Group B. The small sample size precludes any conclusions regarding the efficacy of parsaclisib in patients with relapsed or refractory DLBCL who have previously received ibrutinib.
The efficacy data reported herein are consistent with the findings of a phase 2 study of the pan-PI3K inhibitor, copanlisib (with preferential activity against the α and δ isoforms), conducted in patients with relapsed or refractory DLBCL [Citation22]. The ORR reported herein is 25.5% (N = 55); the ORRs observed with copanlisib in the full cohort and per-protocol analysis sets were 19.4% (N = 67) and 25.0% (N = 40), respectively. The median DOR for copanlisib was 113 days (3.7 months) in the per-protocol population, and was 6.2 months in the current study. Furthermore, parsaclisib and copanlisib demonstrated similar activity in both GCB (ORRs of 18.2% and 13.6%, respectively) and ABC/non-GCB subtypes (31.6% and 37.5%, respectively). Notably, both agents achieved higher response rates in the ABC/non-GCB subtype (copanlisib subtype results are reported in the per-protocol analysis set only). As GCB and ABC/non-GCB subtypes are dependent on the protein kinase B (AKT) and BTK pathways, respectively, and both require PI3K signaling for activity, such broad activity is not surprising [Citation23,Citation24]. By contrast, BTK inhibitors, which block only the BTK pathway, are active primarily in the ABC/non-GCB subtype [Citation24].
Safety results of the current study are similar to those reported in the first-in-human parsaclisib phase 1/2 CITADEL-101 study [Citation19]. The most common AE in our study was rash (multiple terms; 21.8%). Although cutaneous reactions are common with PI3K inhibitors, the frequency and severity of rash in this study is likely confounded by the mandatory PJP prophylaxis. In total, 52 patients (86.7%) received PJP prophylaxis; combination of sulfamethoxazole and trimethoprim, which was the most commonly used PJP prophylactic agent, was administered to 45 patients (75.0%). Notably, the only event of PJP occurred in a patient who had not taken prophylaxis. The majority of diarrhea/colitis reported was grade 1 or 2 (8 of 12 events). One patient each experienced grade 3 colitis and grade 3 enterocolitis; these events occurred during the QD and QW dosing periods, respectively. Grade 3/4 neutropenia was infrequent in the current study (6.7%). Grade ≥3 elevations of AST and ALT were infrequent (reported in 3 and 2 patients, respectively), and the AST/ALT elevation that led to treatment discontinuation was considered related to disease progression rather than to parsaclisib. Grade 3 hyperglycemia occurred in only 1.8% (n = 1) of all patients (no grade 4 event was reported) and there were no reports of grade 3/4 hypertension in our study.
Parsaclisib monotherapy administered QD followed by QW demonstrated clinical activity in patients with relapse or refractory DLBCL. Of note, most patients who experienced disease progression did so within 2 months of treatment, suggesting that tumor progression was not the result of switching to intermittent dosing. Moreover, QD followed by QW parsaclisib dosing was well tolerated in patients with relapsed or refractory DLBCL; no new safety concerns were identified. The parsaclisib QW dosing regimen was administered to 28/55 (51%) patients in Group A; median duration of exposure was 9.0 weeks, and Grade ≥3 TEAEs occurred in 17/28 patients (60.7%), including neutrophil count decreased (10.7%). Despite the high potency of parsaclisib, several toxicities commonly associated with PI3Kδ inhibitors (grade 3/4 diarrhea, colitis, neutropenia, pneumonia, and pneumonitis) were infrequent. Considering that the median time to onset for grade 3/4 diarrhea and colitis is approximately 6 months for parsaclisib administered continuously at high doses [Citation19], the low rate of these AEs in the current study is most likely due to the introduction of an intermittent schedule at week 9. These results are consistent with those in the parsaclisib phase 1/2 CITADEL-101 first-in-human-study [Citation19].
As the study was stopped for futility, further analysis of parsaclisib monotherapy in DLBCL is not supported; however, the observed activity warrants further evaluation in combination with standard therapies and active investigational agents in DLBCL. Trials are underway investigating parsaclisib in combination with rituximab, rituximab plus bendamustine, and ibrutinib (NCT03424122), and bendamustine and obinutuzumab (NCT03039114).
Author contributions
Douglas J. DeMarini was involved in the conception and design of the work. Morton Coleman, David Belada, René-Olivier Casasnovas, Rémy Gressin, Hui-Peng Lee, Amitkumar Mehta, Javier Munoz, Gregor Verhoef, and Keith Fay acquired the data. Claudia Corrado, Douglas J. DeMarini, Wanying Zhao, and Jia Li analyzed the data. All authors drafted the manuscript or revised it critically for important intellectual content.
GLAL-2020-0621-File005.docx
Download MS Word (74.6 KB)Acknowledgments
The authors wish to thank the participants, investigators, and site personnel who participated in this study. Medical writing assistance was provided by Matthew Bidgood, PhD, of Envision Pharma Group (Philadelphia, PA, USA), and funded by Incyte Corporation.
Disclosure statement
Morton Coleman has a consulting or advisory role with Bayer, Celgene, and Gilead; has received research funding from Bayer, Celgene, Gilead, Incyte Corporation, and Merck; owns stock in Kite Pharmaceuticals; and participated in a speakers’ bureau for Bayer, Celgene, Gilead, and Pharmacyclics. David Belada has received research funding or has a consulting role with Celgene, Gilead, Incyte Corporation, Janssen-Cilag, Roche, and Takeda. René-Olivier Casasnovas has a consulting or advisory role with AbbVie, Bristol Myers Squibb, Gilead, Karyopharm, Merck, Roche, and Takeda; and has received research funding from Gilead, Roche, and Takeda. Rémy Gressin reported no potential conflict of interest. Hui-Peng Lee has a consulting or advisory role with Janssen and MSD; and has received an honorarium from Roche. Amitkumar Mehta has a consulting or advisory role with Bristol Myers Squibb, Celgene, Kite, and Spectrum; has participated in a speakers’ bureau for AstraZeneca, Gilead, Kite, Kyowa Kirin, and Spectrum; and has received research funding from Bristol Myers Squibb, Epizyme, Incyte Corporation, Merck, Roche, and Seattle Genetics. Javier Munoz has a consulting role with Alexion, Bayer, BeiGene, Bristol Myers Squibb, Fosun Kite, Gilead/Kite Pharma, Janssen, Juno/Celgene, Innovent, Kyowa, Pfizer, Pharmacyclics, and Seattle Genetics; received research funding from Celgene, Genentech, Incyte Corporation, Janssen, Kite Pharma, Pharmacyclics, Portola, and Seattle Genetics; receives honoraria from Kyowa and Seattle Genetics; and participated in a speakers’ bureau for Acrotech, AstraZeneca, Bayer, BeiGene, Gilead/Kite Pharma, Kyowa, Pharmacyclics, Seattle Genetics, and Verastem. Gregor Verhoef reported no potential conflict of interest. Claudia Corrado, Douglas J. DeMarini, Wanying Zhao, and Jia Li are employees of Incyte Corporation and are stockholders of Incyte Corporation. Keith Fay has received research funding from Incyte Corporation.
Additional information
Funding
References
- Li S, Young KH, Medeiros LJ. Diffuse large B-cell lymphoma. Pathology. 2018;50(1):74–87.
- Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511.
- NCCN Guidelines® B-Cell Lymphomas Version 1.2020. U.S. Plymouth Meeting (PA): National Comprehensive Cancer Network; 2020. https://www.nccn.org/professionals/physician_gls/pdf/b-cell.pdf
- Coiffier B, Sarkozy C. Diffuse large B-cell lymphoma: R-CHOP failure-what to do? Hematology Am Soc Hematol Educ Program. 2016;2016(1):366–378.
- Jurczak W, Długosz-Danecka M, Rivas Navarro F. The rationale for combination therapy in patients with aggressive B-cell non-Hodgkin lymphoma: ten questions. Future Oncol. 2019;15(3):305–317.
- IMBRUVICA® (ibrutinib) capsules, for oral use, tablets, for oral use, U.S. Sunnyvale (CA): Pharmacyclics LLC; 2019.
- Skrabek P, Assouline S, Christofides A, et al. Emerging therapies for the treatment of relapsed or refractory diffuse large B cell lymphoma. Curr Oncol. 2019;26(4):253–265.
- Van Den Neste E, Schmitz N, Mounier N, et al. Outcome of patients with relapsed diffuse large B-cell lymphoma who fail second-line salvage regimens in the International CORAL study. Bone Marrow Transplant. 2016;51(1):51–57.
- Allen JC, Talab F, Slupsky JR. Targeting B-cell receptor signaling in leukemia and lymphoma: how and why? Int J Hematol Oncol. 2016;5(1):37–53.
- Miao Y, Medeiros LJ, Xu-Monette ZY, et al. Dysregulation of cell survival in diffuse large B cell lymphoma: mechanisms and therapeutic targets. Front Oncol. 2019;9:107.
- Valla K, Flowers CR, Koff JL. Targeting the B cell receptor pathway in non-Hodgkin lymphoma. Expert Opin Investig Drugs. 2018;27(6):513–522.
- Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15(1):7–24.
- Puri KD, Gold MR. Selective inhibitors of phosphoinositide 3-kinase delta: modulators of B-cell function with potential for treating autoimmune inflammatory diseases and B-cell malignancies. Front Immunol. 2012;3:256.
- ZYDELIG® (idelalisib) tablets, for oral use. U.S. Foster City (CA): Gilead Sciences, Inc.; 2018.
- COPIKTRA (duvelisib), capsules for oral use. U.S. Needham (MA): Verastem Inc.; 2019.
- ALIQOPA™ (copanlisib) for injection, for intravenous use. U.S. Whippany (NJ): Bayer HealthCare Pharmaceuticals Inc.; 2019.
- Shin N, Koblish H, Covington M, et al. Abstract 2671: INCB050465, a novel PI3Kδ inhibitor, synergizes with PIM protein kinase inhibition to cause tumor regression in a model of DLBCL. Cancer Res. 2015;75(15 Supplement):2671–2671.
- Forero-Torres A, Wertheim MS, Phillips TJ, et al. Abstract CT056: Preliminary safety, efficacy, and pharmacodynamics of a highly selective PI3Kδ inhibitor, INCB050465, in patients with previously treated B-cell malignancies. Cancer Res. 2016;76(14 Supplement):CT056–CT056.
- Forero-Torres A, Ramchandren R, Yacoub A, et al. Parsaclisib, a potent and highly selective PI3Kδ inhibitor, in patients with relapsed or refractory B-cell malignancies. Blood. 2019;133(16):1742–1752.
- Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32(27):3059–3068.
- Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130(16):1800–1808.
- Lenz G, Hawkes E, Verhoef G, et al. Single-agent activity of phosphatidylinositol 3-kinase inhibition with copanlisib in patients with molecularly defined relapsed or refractory diffuse large B-cell lymphoma. Leukemia. 2020;34(8):2184–2197.
- Young RM, Shaffer AL, 3rd, Phelan JD, et al. B-cell receptor signaling in diffuse large B-cell lymphoma. Semin Hematol. 2015;52(2):77–85.
- Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton's tyrosine kinase in B cells and malignancies. Mol Cancer. 2018;17(1):57.