
Abstract
Acute myeloid leukemia (AML) is an aggressive cancer that progresses rapidly with a poor prognosis. Cytogenetic analysis provides the most accurate determination of diagnosis and prognosis however, about 42–48% of AML patients have a cytogenetically normal karyotype. Genetic analysis can provide further information and the identification of new mutations could result in improved risk stratification, prognosis and better understanding of the mechanisms of AML leukaemogenesis. In this study, we analyzed genetic alterations in 16 human AML cases by Haloplex sequencing with confirmation of two previously unreported mutations in the genes DNMT3A and RUNX1 by Sanger sequencing or pyrosequencing. The two novel mutations consist of two frameshift mutations identified in two different AML patients and reported as deleterious by bioinformatic analysis. These mutations confirm the exclusion and co-occurrence of specific gene mutation patterns in AML and may provide further information for patient diagnosis and prognosis.
Introduction
Acute myeloid leukemia (ΑML) is a genetically heterogeneous disease, characterized by the accumulation of genetic alterations in hematopoietic stem and/or progenitor cells. This heterogeneity is reflected by the variable clinical outcomes observed in AML patients. Today, the European Leukemia Net (ELN) genetic risk stratification of AML patients into favorable, intermediate and adverse risk groups is based on cytogenetic abnormalities and certain gene mutations including NPM1, FLT3, CEBPA, RUNX1, TP53, and ASXL1 [Citation1]. The recent inclusion of the above mutations allows the improved classification of AML cases and, in particular, cytogenetically normal AML cases [Citation2]. Cytogenetically normal AML cases account for approximately 50% of AML cases, highlighting the importance of the identification of novel mutations [Citation3].
The aim of this study is to better characterize a set of 16 AML cases using Haloplex targeted sequencing in order to identify novel mutations in genes, which have been implicated in AML, predict their effect on the encoded protein and associate them with clinical, cytogenetic and molecular data of the patients.
Materials and methods
AML patients’ samples and clinical data
DNA from 16 AML patients (six de novo AML, one secondary AML after MDS and nine therapy-related AML after chemotherapy or radiotherapy for a previous malignancy) was obtained from National Center for Research ‘Demokritos’ and Hematology-Lymphomas Department – BMT Unit, Evangelismos Hospital, Athens, Greece with informed consent from each patient. All experimental protocols were approved by Public Health England. It should be noted that the three AML cases after radiotherapy for a previous malignancy could also have had chemotherapy or hormonotherapy but, unfortunately, this information is not available. Clinical data of the patients are provided (). DNA quality was assessed by agarose gel electrophoresis and DNA quantity measured by NanoDrop 2000 (Thermo Fisher Scientific, Paisley, UK).
Table 1. Cytogenetic and clinical data of the sixteen AML patients.
Human AML DNA haloplex sequencing
Human DNA (225 ng) from the 16 patients were sent to Oxford Gene Technology, Begbroke, Oxfordshire for sequencing analysis by Haloplex™ Targeted Enrichment System using the ClearSeq AML panel (Agilent Terchnologies Ltd., Wokingham, UK). Specific AML mutational hotspots analyzed include ASXL1 (exon 12), CSF3R (exons 14,17), CBL (exons 8, 9), CEBPA (exon 1), DNMT3A (exons 4, 8, 13, 15, 16, 18, 19, 20, 22, 23), EZH2 (exons 8, 17, 18), FLT3 (exons 14, 20), IDH1 (exon 4), IDH2 (exon 2), JAK2 (exons 12, 14), MPL (exon 10), NPM1 (exon 11), NRAS (exon 2, 3), RUNX1 (exons 3, 4, 8), SETBP1 (exon 3), SF3B1 (exons 13–15, 17), SRSF2 (exon 1), TET2 (exons 3, 9, 10, 11), TP53 (exons 5–8), U2AF1 (exons 2, 6). Briefly, The DNA was fragmented by restriction enzymes and ClearSeq biotinylated probes specific to the target were hybridized to the fragments forming circular DNA molecules. These circular molecules were separated from the rest of the sample using magnetic streptavidin beads and amplified by PCR ready for MiSeq 150 bp PE Illumina sequencing analysis.
Bioinformatics analysis
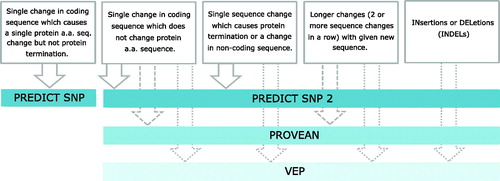
We used our own in house developed pipeline to evaluate different types of genetic modifications effects to protein function (). Nonsynonymous SNP changes were investigated using PredictSNP integrating results from several predictions algorithms, providing a compressed effective evaluation [Citation4]. The sequence changes in gene coding regions not affecting amino acid sequence change (synonymous codons), were investigated by PredictSNP2 [Citation5]. Results are supported by PROVEAN (PROtein Variation Effect ANalyzer) [Citation6] and VEP (Variant Effect Predictor) [Citation7] algorithms. Modifications either located in non-coding regions or causing protein termination without insertion or deletions (INDELS), were analyzed by PROVEAN and VEP. INDELS were evaluated by the VEP algorithm, based on biological rationality and with no computational evaluation performed. The sequence and mutation positions were adjusted to each algorithm based on its reference genome.
Figure 1. Algorithm flow chart. For each type of mutation present, a sequence of different algorithms was used to predict the effect on the protein and organism. The color of algorithm panel represents its effectives. The darker the color, the more effective the evaluation.
Confirmation of novel mutations
Novel mutations were either confirmed by pyrosequencing if primers could be designed for the mutated region or by Sanger sequencing. Briefly, primers for Sanger sequencing confirmation were designed using PrimerQuest software (Integrated DNA Technologies, Leuven, Belgium). PCR reactions consisted of 1 μl 10 X PCR Buffer, 2 μl of 5 × Q solution, 0.25 μl of Taq Polymerase (Qiagen, Manchester, UK), 0.4 μl of each dNTP (Invitrogen/Life Technologies, Carlsbad, USA), 3.15 μl water, 0.5 μl of each primer at 10 μM and 1 μl of DNA. Reactions were performed on a Verti-96 well Thermal Cycler (Applied Biosystems, Foster City, USA) using the following conditions: 94 °C for 3 min, 35 cycles of 30 s at 94 °C, 30 s at 60 °C, 30 s at 72 °C and 72 °C for 10 min. Amplified PCR products were sent to Source Bioscience (Department of Biochemistry, University of Oxford) for Sanger sequencing. Novel mutations were confirmed by pyrosequencing through use of the Pyromark Q48 (Qiagen, UK) according to manufacturer’s guidelines. Primers were designed by the Pyromark Q48 Advanced Software. Reactions consisted of 10 ng DNA, 12.5 μl PyroMark PCR Master Mix 2X, 2.5 μl CoralLoad Concentrate (Red) 10X (Qiagen, UK), 0.5 μl forward primer, 0.5 μl reverse primer (one of the primers to be biotinylated) and RNase-free water to a volume of 25 μl. Reactions were performed on a Verti-96 well Thermal Cycler (Applied Biosystems, USA), using the following conditions: 95 °C for 15 min, 45 cycles of 94 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s. To confirm a strong single band of amplified product, 5 μl PCR product was run on an 1.3% agarose gel. PyroMark Q48 Advanced Reagents were loaded onto the PyroMark with 3 μl Magnetic Beads (Qiagen, UK) and 10 μl PCR reaction.
Results
Amongst the 16 AML patient samples screened for mutations, two novel mutations consisting of frameshift insertions were identified in two different AML patients by bioinformatic analysis (). These novel mutations were predicted to be deleterious using VEP algorithms with these mutations producing a stop codon and termination of the protein. The novel mutations were confirmed by pyrosequencing or sanger sequencing (). Clinical information of the patients with novel mutations are detailed in . All mutations identified in the 16 AML patients by Haloplex sequencing are reported in .
Figure 2. Confirmation of novel mutations. (A) Sanger sequencing confirmation of a Ile715ProfsTer64 mutation (AT->C) in DNMT3A in patient 8, which results in termination of the protein. (B) A Leu144ArgfsTer4 (GGCTGAGC insertion) in RUNX1 in patient 16 was confirmed by pyrosequencing showing a frequency of 28%. An insertion of GCTCAGCC as reported on the reverse strand which corresponds to a GGCTGAGC insertion. Gray shaded regions for pyrosequencing results indicate the region where the presence of the wild-type and mutated change can be detected.
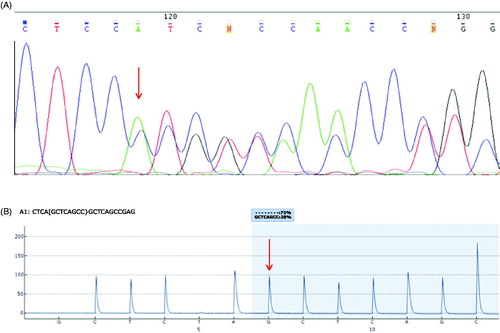
Table 2. Presentation of the two novel mutations detected.
Table 3. Clinical data of AML patients with novel mutations.
Table 4. All genetic mutations reported in all 16 AML patients including previously known mutations and confirmed novel mutations.
Discussion
It is well known that different types of mutations in a gene can lead to changes in the structure of the encoded protein or to a decrease/complete loss of its expression. Mutations can have greater long-term consequences when occurring in hematopoietic stem or progenitor cells when inducing cell growth, inhibiting apoptosis, affecting transcription factors or disrupting normal differentiation, facilitating AML development. The identification of all mutations of the genes implicated in AML prognosis is of great importance as different mutations in the same gene may have different consequences for the AML patient. In this study, we identified two novel human mutations in the genes DNMT3A and RUNX1 in 2 AML patients. The frequencies at which these mutations occurred were reported by both Haloplex sequencing and, when used, pyrosequencing.
DNMT3A is a DNA methyltransferase which is essential for embryonic development and is responsible for the addition of a methyl group to cytosine, forming 5-methylcytosine [Citation8]. DNMT3A is commonly mutated in around 22% of human AML cases, with a lower frequency reported of mutation in Asian populations [Citation9,Citation10]. DNMT3A mutations have been identified as preleukemic mutations arising early in AML evolution and persisting in time of remission [Citation11]. Mutations are most frequently found in the methyltransferase (MTase) region of the gene, where R882 is located [Citation12]. The R882 is the most common mutation, accounting for 60% of all DNMT3A mutations [Citation10,Citation12], leading to reduced DNMT3A enzymatic activity by 50% [Citation9] although no global DNA methylation changes have been detected [Citation12]. The mutation is associated with an up-regulation of HOX family members and HOX cofactor MEIS1 [Citation13,Citation14]. We now report a novel mutation at codon 715 found in patient 8 (). The new mutation is also located in the MTase region. It is present in 40% of the sample cells and is predicted to have a deleterious effect on the protein as it results in its termination. Mutations in DNMT3A have been found to be associated with FLT3, IDH1, IDH2 and NPM1 mutations [Citation10,Citation12,Citation15]. This association is also confirmed in this study as patient 2 also has TET2, IDH2 and NPM1 mutations (). DNMT3A mutations are associated with poor prognostic impact on survival [Citation10,Citation12,Citation16]. Interestingly, patients with DNMT3A mutations have been reported to respond well to the DNA methylation inhibitor decitabine with 75% of DNMT3A-mutated patients achieving complete remission vs 34% of wild type DNMT3A patients, although the size of that study was small at just 46 patients [Citation17]. In this case, however, the patient is still alive more than 84 months post diagnosis of AML. This surprisingly long survival could be mainly due to the bone marrow transplantation, but NPM1 mutation which has been associated with favorable survival [Citation18] and the novel DNMT3A mutation found in this study could have also contributed.
Another mutation found in our samples was in RUNX1 gene, also known as acute myeloid leukemia 1 protein (AML1). It is a transcription factor activating genes involved in hematopoiesis and is essential in early development with homozygous mutations in mice resulting in embryonic death [Citation19]. RUNX1 mutations have been frequently reported in MDS patients, and also in AML cases. A higher frequency of RUNX1 mutations was reported in cases which have been developed from MDS (24%), rather than in de novo AML cases (9%) [Citation20]. In our study, the novel RUNX1 mutation at codon 144 () is located in exon 8 in the transactivation domain which binds with various growth factors, signaling molecules and transcription activators (). The most common types of mutations are frameshift mutations [Citation20,Citation21]. This novel RUNX1 mutation is present in 22% of the sample as measured by Haloplex sequencing, leading to termination of the protein. Pyrosequencing analysis of the RUNX1 frameshift at codon 144 found a similar frequency of the mutation at 28% of the sample cells (). Interestingly, RUNX1 mutations which have shown to be significantly associated with ASXL1 mutations [Citation20], were confirmed in both patient 5 and patient 16 in this study (). Mutual exclusivity was also seen in patients that had RUNX1 mutations and those that had FLT3 and NPM1 mutations, as also previously reported [Citation15]. Overall survival is significantly lower in patients with a RUNX1 mutation [Citation20–22]. In this study, 16 with the novel RUNX1 mutation relapsed and died of resistant disease at 35 months, confirming the adverse prognosis of this mutation.
Another novel mutation was identified and confirmed in the gene SRSF2. A point mutation at codon 96 in patient 10 changed proline to leucine, predicting to have a deleterious effect on the protein (). SRSF2 is a splicing factor [Citation23] most commonly mutated in chronic myelomonocytic leukemia (CMML) or MDS cases, rather than AML cases [Citation24]. Spliceosome genes are found to be mutated in 5% of AML patients, with mutations in SRSF2 found in only 1% of AML patients [Citation24,Citation25]. Therefore, mutations in SRSF2 are rare in AML and their prognosis is currently unknown. Frequent persistence of SRSF2 mutations has been found in intensively treated AML patients in first complete remission [Citation26]. The most common point mutation is at codon Pro95 [Citation27], which is located in the RNA recognition motif forming a bond with RNA [Citation28]. The novel mutation reported in patient 10 of this study occurs at Pro96 at a frequency of 23% with a predicted strong deleterious effect, possibly interfering with RNA binding. Pyrosequencing analysis has found the mutation to occur at a higher level in 48% of the sample cells (). This patient was diagnosed with AML following chemotherapy treatment for MDS; this is in line with the observation that SRSF2 mutations are found especially in secondary AML patients [Citation29]. Unfortunately, we do not have further information on relapse and survival for this patient. Also, although a new mutation, this mutation is just one amino acid downstream of the common Pro95 hotspot and so it is unlikely that this mutation identifies a new mechanism of gene disruption.
Figure 3. Additional mutations of interest. (A) A Pro96Leu mutation (C->T) in SRSF2 in patient 10 was confirmed by pyrosequencing showing a frequency of 48%. (B) Sanger sequencing confirmation of the Gln821Ter (CAG->TAG) TET2 mutation in patient 5 which results in termination of the protein.
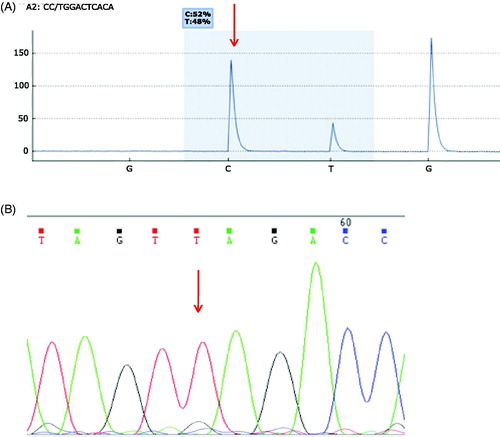
A mutation in the gene TET2 was also reported at Gln821 which, although previously reported in myeloproliferative neoplasms (MPNs) [Citation30], has not been reported in AML before. This is of interest as the same genetic change may lead to a different prognosis in different hematological malignancies. TET proteins function in DNA methylation by converting 5-methyl-cytosine to 5-hyroxymethylcytosine (5hmC) [Citation31]. TET2 mutations have been identified in a range of haematological malignancies such as AML, CML and MDS [Citation32]. These mutations occur in 8–28% of adult AML cases [Citation15,Citation33,Citation34]. TET2 mutations are located all along the gene, most of which are single nucleotide mutations, found to result in loss of function of generating 5hmC [Citation16]. The Gln821Ter mutation found in our study () was present in 95% of the bone marrow sample by Haloplex sequencing and, confirmed by Sanger sequencing, where a clear, high mutational peak was observed (T) with a very small wild-type peak (G). This mutation was identified in patient 5 where 70% of bone marrow cells were blasted. This frequency suggests that TET2 Gln821Ter probably occurred early in leukaemogenesis and is likely to have been a driver mutation. The slight discrepancy between the percentage of TET2 mutation found in 95% of the sample, while the blasts of the patients were lower (70%) is in line with previous studies which have reported that TET2 mutations are associated with clonal hematopoiesis in healthy elderly people frequently increased with age and are early events in leukemogenesis [Citation29]. This patients’ leukemic cells also contained mutations in RUNX1, JAK2 and ASXL1 genes. The role of a TET2 mutation on patient survival is unclear. There are conflicting reports with some studies reporting that it can significantly affect the overall survival in AML patients [Citation16,Citation34], particularly in cases with an intermediate prognosis karyotype [Citation33], while in other studies it has been shown that there is no significant difference in survival [Citation35]. In this case, the patient unfortunately died of resistant disease, possibly related to this TET2 mutation found in 95% of the sample or to the combination of his mutations. Comparing with other cases with this particular mutation would be of interest.
In summary, we have identified 2 novel mutations, two frameshift mutations in the genes RUNX1 from a ->GGCTGAGC insertion (Leu144ArgfsTer4) and in DNMT3A from an AT->C deletion and insertion (Ile715ProfsTer64), both of which predict a deleterious effect on the encoded protein with the termination of the protein. The identification of novel gene mutations and prediction of their respective effects on the encoded proteins reported in this study provides useful information bringing new light into the complexity of the genetic landscape of AML, confirming exclusion and co-occurrence of specific gene mutation patterns. The evaluation of their prognostic significance may contribute to a better assignment of patients to AML diagnostic and prognostic categories, and perhaps to a personalized treatment in the future by identifying new therapeutic targets hence improving clinical course and survival of AML patients.
Ethics approval
The project was in accordance with the declaration of Helsinki. The Ethical Committee of the NSCR ‘Demokritos’ allows the use of anonymous or encoded residual diagnostic samples for research purposes when informed consent was provided from patients. Informed consent was provided from patients and donors enrolled in the study.
Author contributions
Conceptualization, G.O.B., C.B., K.M. and M.P.; Formal Analysis, G.O.B, J.Z. and J.P.; Writing – Original Draft Preparation, G.O.B.; Writing – Review & Editing, C.B., K.M, M.P., J.Z. and J.P.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Dohner H, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447.
- Kayser S, Levis MJ. Clinical implications of molecular markers in acute myeloid leukemia. Eur J Haematol. 2019;102(1):20–35.
- Dohner K, Dohner H. Molecular characterization of acute myeloid leukemia. Haematologica. 2008;93(7):976–982.
- Bendl J, Stourac J, Salanda O, et al. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol. 2014;10(1):e1003440.
- Bendl J, Musil M, Štourač J, et al. PredictSNP2: a unified platform for accurately evaluating SNP effects by exploiting the different characteristics of variants in distinct genomic regions. PLoS Comput Biol. 2016;12(5):e1004962.
- Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745–2747.
- McLaren W, Gil L, Hunt SE, et al. The ensembl variant effect predictor. Genome Biol. 2016;17(1):122.
- Okano M, Bell DW, Haber DA, et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257.
- Yamashita Y, Yuan J, Suetake I, et al. Array-based genomic resequencing of human leukemia. Oncogene. 2010;29(25):3723–3731.
- Hou H-A, Kuo Y-Y, Liu C-Y, et al. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 2012;119(2):559–568.
- Medinger M, Passweg JR. Acute myeloid leukaemia genomics. Br J Haematol. 2017;179(4):530–542.
- Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–2433.
- Yan X-J, Xu J, Gu Z-H, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43(4):309–315.
- Ferreira HJ, Heyn H, Vizoso M, et al. DNMT3A mutations mediate the epigenetic reactivation of the leukemogenic factor MEIS1 in acute myeloid leukemia. Oncogene. 2016;35(23):3079–3082.
- The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074.
- Aslanyan MG, et al. Clinical and biological impact of TET2 mutations and expression in younger adult AML patients treated within the EORTC/GIMEMA AML-12 clinical trial. Ann Hematol. 2014;93(8):1401–1412.
- Metzeler KH, Walker A, Geyer S, et al. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia. 2012;26(5):1106–1107.
- Dohner K, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106(12):3740–3746.
- Okuda T, van Deursen J, Hiebert SW, et al. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84(2):321–330.
- Gaidzik VI, Teleanu V, Papaemmanuil E, et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia. 2016;30(11):2282.
- Schnittger S, Dicker F, Kern W, et al. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood. 2011;117(8):2348–2357.
- Jalili M, et al. Prognostic value of RUNX1 mutations in AML: a meta-analysis. Asian Pac J Cancer Prev. 2018;19(2):325–329.
- Krainer AR, Conway GC, Kozak D. Purification and characterization of pre-mRNA splicing factor SF2 from HeLa cells. Genes Dev. 1990;4(7):1158–1171.
- Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–69.
- Kihara R, Nagata Y, Kiyoi H, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia. 2014;28(8):1586–1595.
- Rothenberg-Thurley M, Amler S, Goerlich D, et al. Persistence of pre-leukemic clones during first remission and risk of relapse in acute myeloid leukemia. Leukemia. 2018;32(7):1598–1608.
- Arbab Jafari P, Ayatollahi H, Sadeghi R, et al. Prognostic significance of SRSF2 mutations in myelodysplastic syndromes and chronic myelomonocytic leukemia: a meta-analysis. Hematology. 2018;23(10):778–784.
- Daubner GM, Cléry A, Jayne S, et al. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. Embo J. 2012;31(1):162–174.
- Bullinger L, Dohner K, Dohner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol. 2017;35(9):934–946.
- Grinfeld J, Nangalia J, Baxter EJ, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018;379(15):1416–1430.
- Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935.
- Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–2301.
- Chou W-C, Chou S-C, Liu C-Y, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118(14):3803–3810.
- Gaidzik VI, Paschka P, Späth D, et al. TET2 mutations in acute myeloid leukemia (AML): results from a comprehensive genetic and clinical analysis of the AML study group. J Cin Oncol. 2012;30(12):1350–1357.
- Weissmann S, Alpermann T, Grossmann V, et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. 2012;26(5):934–942.