
Abstract
IgD multiple myeloma is uncommon. Patients generally present at a younger age and have shorter progression free and overall survivals (OSs). Its rarity has inhibited development of a specific risk stratification system or informed best treatment protocols. We present interphase fluorescence in situ hybridization results from a group of 29 cases. These showed evidence of a decreased male to female ratio, decreased OS in patients aged 70 and over, better outcomes in those with kappa light chain restriction, and CD56 positive patients had longer survivals than those lacking CD56. We discuss the biology of IgD multiple myeloma, the need for prospective studies, and challenges for improvements in diagnosis and treatment. We suggest an International Register to accelerate development of best practice guidelines for diagnosis, risk stratification, and treatment.
Introduction
Immunoglobulin D-secreting multiple myeloma (IgD MM) is a rare form of the disease, accounting for approximately 1–2% of cases. Its frequency is possibly underestimated because of a protein immunochemistry workup lacking full discretionary reflex testing. Interestingly, UK NEQAS IgD MM samples have not been circulated since 2012, a factor presumably related to the paucity of suitable samples. Furthermore, only ∼50% of participating laboratories correctly identified or confirmed the presence of IgD M-protein, highlighting the urgent need for improvements in early diagnosis, particularly pertaining to laboratory methodological and interpretive performance. This in turn may contribute to improved overall survivals (OSs).
Although it shares many characteristics with other MM isotypes, patients are reported to present at a younger age and have shorter progression-free survival (PFS) and OS. Morris et al. reviewed over 20,000 myeloma patients receiving autologous stem cell transplants (ASCTs) between 1986 and 2007, showing 379 IgD MM patients with a median OS of 43.5 months compared to 62.3 months for IgG, IgA, and light chain (LC) MM, despite higher rates of complete remission. As with other myeloma isotypes, there is a small group of long-term survivors [Citation1].
In the recent Collaboration to collect Autologous transplant outcomes in Lymphoma and Myeloma (CALM) study conducted by the European society of Blood and Marrow Transplantation (EBMT) on treatment, including the effects of mobilizing stem cells with plerixafor, Lawless et al. identified 29 IgD MM patients in a total of 2803 newly diagnosed MM cases [Citation2]. These IgD patients were younger at presentation, with median age at first autograft 55.6 years vs. 59.5 years, later ISS stage, poorer stem cell mobilization, and shorter OS in comparison to other isotypes (48.5 months vs. 81.7 months) [Citation2].
Although there is a lack of concordance between studies, cytogenetic aberrations in IgD MM suggest a distinct pattern. As with MM overall, application of interphase fluorescence in situ hybridization (iFISH) detects abnormalities in most cases [Citation3,Citation4]. The CCND1:;IGH translocation t(11;14) is more frequently observed in IgD MM than in IgG, IgA, and LC MM [Citation5,Citation6]. Interestingly, this translocation does not confer poor prognosis in MM overall [Citation7]. Studies also showed an association between t(11;14) and a CD56 negative phenotype [Citation5,Citation6]. An earlier study associated t(11;14) with IgM, IgE, and non-secretory MM [Citation8]. Additionally, the 1q21 amplification, observed with greater frequency in late disease, has been shown to be more common in IgD MM [Citation9], possibly due to more advanced disease at diagnosis [Citation10]. Interestingly, we have found no clear-cut evidence reported in the literature of increased frequency of del(17p), or TP53 mutations, in IgD MM ().
Table 1. Acquired cytogenetic aberrations.
We present analysis of results of a total of 3198 newly presenting MM patients diagnosed over a 14-year period, of which 29 had IgD MM. We discuss the limitations of retrospective studies, the need for well-designed prospective data collection, and possible factors contributing to poorer outcomes in IgD MM patients, including the biology of the disease, inaccurate and/or late diagnosis, and prospects for improved treatment modalities.
Patients, materials, and methods
Patients
We studied iFISH results and other data on 29 IgD MM patients, identified in a cohort of 3198 newly diagnosed MM patients presenting between 2005 and 2018. Data are from the UK population-based Haematological Malignancy Research Network (HMRN; www.hmrn.org) which, with a catchment population of nearly 4 million people, has a socio-demographic composition that broadly mirrors that of the UK overall. Initiated in 2004, full details of its structure, data collection methods and ethical approvals have been described previously [Citation12]. Briefly, within HMRN, patient care is provided by 14 hospitals, organized into five multi-disciplinary teams (MDTs); and clinical practice adheres to national guidelines. As a matter of policy, all diagnoses across the HMRN region are made to current WHO diagnostic criteria and coded by clinical specialists at a single integrated haematopathology laboratory – the Haematological Malignancy Diagnostic Service (www.hmds.info).
The 29 IgD MM patients identified, representing 0.9% of the MM cohort, was lower than the 1–2% reported in the literature. A possible reason for this may be under detection, in parallel with over detection of LC MM for reasons explained below, as serum protein electrophoresis was carried out in the UKNHS laboratories of the referring hospitals, prior to bone marrow (BM) aspirate and trephine biopsy analyses in the centralized specialist laboratory.
Materials and methods
The iFISH technique underwent continuous development during the 14-year timespan of the analysis, carried out initially on BM aspirate smears, and eventually CD138 selected plasma cells from 2011 onwards. Furthermore, the quality and range of the iFISH probes gradually improved during the reported period, leading to better accuracy and precision as patterns of sample handling evolved.
As BM aspirate samples needed to be processed for CD138 plasma cell selection prior to confirmed diagnosis, the decision to carry out plasma cell selection was based on the flow cytometry results. All BM samples with greater than 90% neoplastic plasma cells identified in the total plasma cell population underwent plasma cell selection. Approximately, 5% of samples had either insufficient sample to proceed or showed a selection purity of <50% plasma cells (usually due to sample degradation during transport to the laboratory). The purified plasma cells were fixed in suspension in methanol acetic acid fixative (Carnoy’s solution) and stored in suspension at –20 °C. Once a diagnosis of myeloma was confirmed, the sample had FISH testing carried out using a panel of commercial probes, as described by O'Connor et al. [Citation13].
Patient age, sex, LC restriction, CD56 status, and OS were noted when available and median OS calculated.
Results
Twenty-nine IgD patients were identified, of whom 15 were female and 14 male, while 20 were CD56 positive and nine were CD56 negative ( and ). Light chain restriction was available on only 14 patients and of these, three expressed kappa and 11 expressed lambda LCs. Eight of the 29 had no iFISH results, either because of unsuccessful test outcome (N = 5), mainly due to inadequate sample, or presentation before iFISH was introduced routinely in 2005 (N = 3).
Table 2. Summary statistics of IgD MM patients.
Table 3. Median overall survival in patient subgroups (where outcome was known).
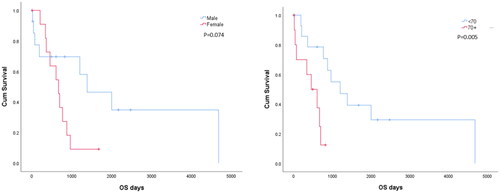
Analysis of outcomes as defined by median OS showed better survivals associated with: age at presentation, with under 70-years old patients surviving longer than those who were aged 70 and over; sex where males survived longer than females; LC restriction with kappa expressing surviving longer than those who showed lambda LC restriction; and CD56 expression where positive patients survived longer that those where CD56 expression was absent (). Sample Kaplan–Meier’s survival curves were plotted and compared using the log rank test ().
Figure 1. Kaplan–Meier’s survival plots in patient subgroups.
There was no significant pattern of acquired cytogenetic aberration identified in the IgD MM patients. We failed to identify any increase in the frequency of t(11;14), del17p or aberrations involving 1q or 1p. The only suggestion of a distinct pattern was that of MYC rearrangement which was found in 4/14 IgD MM patients (28%), compared to 91/1646 IgG MM patients (5.5%). However, the number of IgD patients was small, so this needs to be interpreted with caution but warrants investigation of a larger cohort of patients, or perhaps a meta-analysis of several large studies. Furthermore, some probes used did not allow identification of a partner chromosome.
Discussion
These analyses were restricted by patchy availability of data. They emphasize the difficulty in interpreting, retrospectively, large datasets of diagnostic results, with many different hospitals using the service, leading to incomplete data when some are not provided by the referring hospital. Evolving methodologies and reagents, particularly improved iFISH probes, and data handling systems which were not designed for easy integration of test results, can contribute to inferior analytical capabilities than those achieved by well-designed prospective studies.
Factors contributing to poorer outcomes in IgD MM patients, compared to MM overall, include delayed diagnosis or failure to correctly identify the IgD M-protein, and differences in the biology of the IgD producing MM cell, compared to that of the other M-protein isotypes. The detection and identification of these other M-protein isotypes, IgG, IgA, and IgM, is facilitated by routine electrophoretic and immunochemical analyses because of their relative abundance in normal serum and the typically elevated levels of the involved Ig in MM. However, an IgD M-protein is more challenging to detect as it is not measured as part of an Ig panel and, as a consequence of its low synthetic and high catabolic rates, the M-protein may be below the detection capability of current electrophoretic techniques [Citation2,Citation5]. Also, when a M-protein is isotyped using the standard immunofixation electrophoresis (IFE) panel, the presence of a band with LC antisera (free and bound) can be misinterpreted as a monoclonal LC gammopathy.
The degree of this type of misidentification was reflected in the above-mentioned UK NEQAS Monoclonal Scheme EQA distribution (2012) which contained an IgD M-protein, measuring approximately 8 g/L. Seventy-five laboratories (23%) reported LC only and did not consider the possibility of an intact IgD M-protein, representing no improvement in identification since 2007. Furthermore, we suspect that this failure to correctly identify all the IgD MM patients contributed to the lower-than-expected IgD numbers in our patient cohort as the protein workup took place in the participating hospital laboratories, rather than in a centralized specialized laboratory. The laboratory workup is critically important and should include discretionary reflex IFE and serum free light chain (sFLC) measurement.
IgM is the only immunoglobulin (Ig) isotype expressed by normal, immature B cells. IgD expression commences when B cells exit the BM to populate peripheral lymphoid tissues [Citation14]. On maturation, but usually before antigen presentation, B-cells show transmembrane expression of both IgM and IgD with identical specificity while functioning as a B-cell receptor (BCR) for antigen recognition prior to class switching [Citation15,Citation16]. IgD signaling is only triggered by repetitive multivalent immunogens, whereas IgM can be triggered either by soluble monomeric or multivalent immunogens [Citation14]. IgM expression is lost as IgM+IgD+ B cells differentiate into IgD plasma cells [Citation16].
It is reasonable to hypothesize that IgD MM may be characterized by an end stage B cell which has failed to undergo Ig class switching on antigen presentation, produces Ig in quantities more representative of earlier B lineage cells, and may even have failed to undergo IGHV somatic hypermutation. Although sporadic MM studies have failed to identify any commonality of IGHV gene usage, as found in chronic lymphocytic leukemia (CLL), and have shown evidence of somatic hypermutation of IGHV genes, we have not found any reports in the literature of IGHV somatic hypermutation and gene usage studies in IgD MM. Such a study could contribute to a better understanding of the biology of this uncommon form of MM, perhaps helping to explain the poorer outcomes.
Although ASCT has improved survivals in IgD MM, they do not reach those attained in MM overall. The use of more targeted therapies, including novel agents, may contribute to improved outcomes. For example, the reported efficacy of venetoclax-based regimens in relapsed/refractory t(11;14) MM is of particular interest in the context of being the first anti-MM agent with a reliable biomarker, with t(11;14) predicting efficacy. Translocations involving chromosome 14 are a recurrent finding in MM and approximately 15% of patients demonstrate a t(11;14) (q13;q32) involving the CCND1::IGH genes. Although not confirmed in our study, IgD MM patients have been reported to demonstrate a higher incidence of t(11;14) than in MM overall. This juxtaposition results in CCND1 being overexpressed, leading to kinase activation and tumor cell proliferation. t(11;14) cases in MM are predicted to be BCL-2-dependent, resulting in upregulation of antiapoptotic proteins and thereby making BCL-2 a potential target in this subtype of myeloma [Citation17].
Venetoclax is a BCL-2 inhibitor and promotes apoptosis via a TP53 mutation-independent pathway and is of proven efficacy in CLL patients with del(17p) and/or TP53 mutation [Citation18]. It has also been demonstrated to cross the blood brain barrier in CLL and is therefore of potential efficacy in central nervous system MM (CNS-MM) [Citation19,Citation20]. Several phase III trials are currently underway employing venetoclax in patients with relapsed refractory MM (RRMM). With the reported increased frequency of t(11;14) in IgD MM, it may have the potential to contribute to improved outcomes and be the forerunner of other biomarkers with potential to predict responsiveness to particular therapies [Citation21,Citation22].
Given the relatively uncommon frequency of IgD MM, and limited numbers of cases included in clinical trials, an International Register of such cases may be the best way to identify optimum diagnostic, risk stratification, and treatment strategies for this particularly problematic form of MM affecting younger patients and associated with poorer survivals.
Author contributions
PAE and SJMO’C contributed equally to this paper and are joint first authors. SJMO’C developed and provided oversight of the iFISH analyses, contributed and analyzed the data from the regional diagnostic center, and contributed to initial drafts and final edit of the manuscript. PAE wrote the initial drafts of the manuscript, carried out statistical analysis, and contributed to the overall document and final edit. WID provided major input on the diagnosis of IgD MM, and to the initial drafts and final edit of the manuscript. CEMcC, PTE, and TCMM provided clinical data, including treatment strategies, and contributed to the initial drafts and final edit of the manuscript. HDA acts as CI for the plasma cell dyscrasia study at Altnagelvin Area Hospital, supervised the collaboration with SJMO’C, co-wrote the manuscript with PAE, and co-ordinated the contributions of the other coauthors and final edit.
Acknowledgements
The authors would like to thank Dan Painter, Epidemiology and Cancer Statistics Group, York University.
Disclosure statement
The authors declare no conflicts of interest.
Additional information
Funding
References
- Morris C, Drake M, Apperley J, et al. Efficacy and outcome of autologous transplantation in rare myelomas. Haematologica. 2010;95(12):2126–2133.
- Lawless S, Sbianchi G, Morris C, et al. IgD subtype but not IgM or non-secretory is a prognostic marker for poor survival following autologous hematopoietic cell transplantation in multiple myeloma. Results from the EBMT CALM (collaboration to collect autologous transplant outcomes in lymphomas and myeloma) study. Clin Lymphoma Myeloma Leuk. 2021;21(10):686–693.
- Chen L, Fan F, Deng J, et al. Clinical characteristics and prognosis of immunoglobulin D myeloma in the novel agent era. Ann Hematol. 2019;98(4):963–970.
- Selene II, Jose JA, Khalil MJ, et al. Presentation patterns, diagnostic markers, management strategies, and outcomes of IgD multiple myeloma: a systematic review of literature. Cureus. 2019;11(2):e4011.
- An G, Xu Y, Shi L, et al. t(11;14) multiple myeloma: a subtype associated with distinct immunological features, immunophenotypic characteristics but divergent outcome. Leuk Res. 2013;37(10):1251–1257.
- Sun Q, An G, Liu E, et al. The clinic and pathologic significance of plasma cell myeloma with CCND1. Zhonghua Xue Ye Xue Za Zhi. 2015;36(9):775–779.
- Sonneveld P, Avet-Loiseau H, Lonial S, et al. Treatment of multiple myeloma with high-risk cytogenetics: a consensus of the International Myeloma Working Group. Blood. 2016;127(24):2955–2962.
- Avet-Loiseau H, Garand R, Lode L, et al. Translocation t(11;14)(q13;q32) is the hallmark of IgM, IgE, and nonsecretory multiple myeloma variants. Blood. 2003;101(4):1570–1571.
- Lu J, Lu J, Chen W, et al. Clinical features and treatment outcome in newly diagnosed Chinese patients with multiple myeloma: results of a multicenter analysis. Blood Cancer J. 2014;4:e239.
- Sawyer JR. The prognostic significance of cytogenetics and molecular profiling in multiple myeloma. Cancer Genet. 2011;204(1):3–12.
- Gao XY, Ma YP, Chao Y, et al. Clinical characteristics and survival analysis of patients with IgD multiple myeloma. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2021;29(2):547–552.
- Smith A, Howell D, Crouch S, et al. Cohort profile: the Haematological Malignancy Research Network (HMRN): a UK population-based patient cohort. Int J Epidemiol. 2018;47(3):700–700g.
- O'Connor SJM, Turner KR, Barrans SL. Chapter 3: practical applications of fluorescent in situ hybridization techniques in clinical diagnostic laboratories. In: Nielsen B S ' Jones J editors. In situ hybridization protocols. 5th ed., City: Totowa, NJ 07512, USA:Humana Press; 2020
- Übelhart R, Werner M, Jumaa H. Assembly and function of the precursor B-cell receptor. Curr Top Microbiol Immunol. 2016;393:3–25.
- Cooper MD. The early history of B cells. Nat Rev Immunol. 2015;15(3):191–197.
- Gutzeit C, Chen K, Cerutti A. The enigmatic function of IgD: some answers at last. Eur J Immunol. 2018;48(7):1101–1113.
- Pistofidis R, Ghobrial I. Targeting a myeloma translocation for the first time: the t(11;14) journey. Hematologist. 2018;15(4).
- Campo E, Cymbalista F, Ghia P, et al. TP53 aberrations in chronic lymphocytic leukemia: an overview of the clinical implications of improved diagnostics. Haematologica. 2018;103(12):1956–1968.
- Egan PA, Elder PT, Deighan WI, et al. Multiple myeloma with central nervous system relapse. Haematologica. 2020;105(7):1780–1790.
- Reda G, Cassin R, Dovrtelova G, et al. Venetoclax penetrates in cerebrospinal fluid and may be effective in chronic lymphocytic leukemia with central nervous system involvement. Haematologica. 2019;104(5):e222–e223.
- Jelinek T. Venetoclax: the first anti-myeloma agent with a reliable biomarker. Br J Haematol. 2020;189(6):1003–1005.
- Basali D, Chakraborty R, Rybicki L, et al. Real-world data on safety and efficacy of venetoclax-based regimens in relapsed/refractory t(11;14) multiple myeloma. Br J Haematol. 2020;189(6):1136–1140.