
Abstract
Gene expression profiles of blood can reflect the physiopathologic status of the immune system. The dynamic microRNA (miRNA) expression profiles of peripheral blood from pigs at different developmental stages, and how differential expression of miRNAs might relate to immune system development, are unknown. In this study, peripheral blood samples taken at five developmental stages were used to construct 15 miRNA libraries (three biological replicates/stage): 0 days (newborn), 30 days (weaning), 60 days (weaned), and 180 and 360 days (puberty). We identified 295 known mature miRNAs. Hierarchical clustering of the miRNA expression profile showed significant differences between individuals at the neonatal and postnatal stages. Functional enrichment analysis revealed that miRNAs differentially expressed between pairwise comparisons of the developmental stages were over-represented in immune-related pathways such as toll-like receptor signaling. The time-course of expression of the over-representated miRNAs exhibited a pattern of steady decline over time, for both the complete miRNA compendium and immune-related miRNAs. We identified six marker miRNAs that were highly negatively correlated with chronologic age and enriched for genes involved in immune-related pathways. This study of a peripheral blood miRNA transcriptome offers insight into immune system development in swine and provides a resource for pig genome annotation.
Keywords:
Introduction
Pigs, which were one of the earliest animals to be domesticated, play an important role in the agricultural economy. The high mortality of piglets has caused huge economic losses for the pig livestock industry. Among many factors that affect mortality in newborn piglets, a developing immune system that lacks immunologic memory is the most important. From late gestation to weaning, adaptive immunity in newborns gradually replaces the protection provided by passive immunity and maternal antibodies.Citation1,Citation2 This development of adaptive immunity continues until puberty.Citation1 During the critical stages of immune development, young piglets are easily infected by pathogens including viruses. Compared with adult pigs, young piglets are more susceptible to porcine reproductive and respiratory syndrome virus (PRRSV) infection.Citation3 The mortality rate of newborn piglets (<7 days old) infected with porcine epidemic diarrhea virus (PEDV) approaches 100%.Citation4–6 Therefore, a better understanding of how the pig immune system develops after birth is needed.
In mammals, blood contains various cells of the immune system, including T cells, B cells, monocytes and granulocytesCitation7 and plays an important role in transporting nutrients and immune cell to required sites. Gene expression profiles in blood can reflect the status of the immune system and provide a snapshot of immune regulatory networks.Citation7,Citation8 Therefore, we chose peripheral blood as the source material for our analysis.
MicroRNA (miRNA) comprises a group of small non-coding RNAs (generally 19–24 nucleotides [nt] in length) that repress protein expression and promote the degradation of target mRNA by binding to the 3′-untranslated region of target mRNA. Several miRNAs have been confirmed to play functional roles in the immune response, including immune cell differention, and activation and regulation of the immune response to infection.Citation7 Two such miRNAs, miR-150 and miR-155, are important for B cell development;Citation9,Citation10 miR-155 is also known for regulation of antibody production and other functions of B and T lymphocytes.Citation9 MiR-146a may negatively regulate the nuclear factor (NF)-κB signaling pathway by targeting tumor necrosis factor receptor-associated factor 6 (TRAF6) and interleukin-1 receptor-associated kinase (IRAK1), which control innate immune signaling in response to lipopolysaccharide.Citation11 MiR-221-5p inhibits PEDV replication by directly targeting the 3′-untranslated region of the viral genome and activating the NF-κB-signaling pathway,Citation12 while miR-26a suppresses PRRSV replication by activating innate immune responses such as the type I interferon signaling pathway.Citation13 Therefore, miRNAs may play a critical role in immune system development.
A dynamic profile of peripheral blood miRNA expression across postnatal development has not been extensively studied in pigs but could reveal relationships between differential expression of miRNAs and development of the immune system. To fill this gap, we performed small-RNA sequencing on peripheral blood samples from pigs drawn during five critical stages of postnatal development: newborn (0 days), weaning (30 days), weaned (30 days), and puberty (180 and 360 days).
Materials and methods
Ethics statement and sample collection
Peripheral blood was obtained from healthy female Tibetan pigs at the ages of 0 (Z0), 30 (Z30), 60 (Z60), 180 (Z180) and 360 (Z360) days after birth; three individuals were sampled for each time point. All samples were immediately frozen in liquid nitrogen and stored at −80 °C until subsequent RNA isolation. All experimental protocols were approved by the Institutional Animal Care and Use Committee of Sichuan Animal Science Academy (permit number: XKY-S20140604).
Small-RNA library construction and sequencing
Total RNA was extracted from peripheral blood using Trizol (Ambion, Carlsbad, CA. USA) according to the manufacturer’s protocol. A NanoDrop ND-2000 spectrophotometer (Thermo NanoDrop Technologies, Wilmington, DE, USA) and Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA) were both used to detect the integrity and concentration of RNA samples. The average optical density of each sample at 260/280 nm was 1.90 (1.90 ± 0.12, n = 15) and the average RNA integrity number of each sample was 9.60 (9.60 ± 0.33, n = 15; Table S1). A small-RNA Sample Pre Kit was utilized to build cDNA libraries, which were initially quantified with a Qubit 2.0 fluorometer. The Agilent Bioanalyzer 2100 was then used to identify libraries with the required insert size. Quantitative PCR was used to accurately quantify the effective concentration (>2 nM) to ensure the quality of the library. Finally, each library was sequenced on an Illumina HiSeq™ 2500 platform.
Read mapping and miRNA quantification
To obtain clean reads, those with >10% unidentified nucleotides, >10 nucleotides in length aligned to the adapter (allowing ≤10% mismatches), and >50% of bases with a Phred quality score <5 were filtered. Clean reads were then aligned to the pig genome (Sus scrofa; Sscrofa 11.1) using Bowtie (version 1.1.1) and only mapped reads were retained. To quantify known porcine miRNAs, all retained clean reads were searched against miRbase (version 21; http://www.mirbase.org/). The tools miRDeep2 and miREvo were further applied to predict and quantify novel miRNAs. Considering the reliability of miRNA, miRNAs with a read count ≥1 in at least one replicate of each sample were retained.
Short time-series expression miner (STEM) analysis
We used STEM softwareCitation14 to identify and visualize over-represented temporal miRNA expression profile patterns across different developmental stages. The clustering algorithm of STEM first selects a group of potential temporal model profiles, then assigns each miRNA to the best representative model profile based on the correlation coefficient. A permutation test was then used to estimate the number of miRNAs assigned to each model profile and was repeated for a large number of permutations of the time points. The average number of miRNAs assigned to the best representative model profile in all permutation runs was used as the number of expected miRNAs assigned to the model profile. The significance of the number of miRNAs assigned to a model profile, compared with the expected number, was computed using a standard hypothesis test.Citation15
Identification of differentially expressed (DE) miRNAs and functional enrichment analysis
We identified DE miRNAs between different time points using DESeq2 package.Citation16 The miRNAs with a > 1.3-fold change and an adjusted p value < 0.05 were identified as DE miRNAs.
We used Basic Local Alignment Search Tool (BLAST, version 2.9.0) to convert pig miRNAs to their human orthologues, then performed functional enrichment analyses on the human orthologues utilizing DIANA-miRPath (http://www.microrna.gr/miRPathv3).Citation17 In short, we carried out all-against-all BLAST analysis (settings: E-value, 1 × 10−5; word_size, 7; strand, plus; other parameters, default) on human and pig miRNA precursor sequences; reciprocal best hits were retained as single-copy orthologues. Human and pig miRNA precursor sequences were downloaded from miRbase (version 21).Citation18 We further converted pig-to-human orthologous precursor miRNA pairs to orthologous mature miRNA pairs, by matching the 5p (or 3p) pig mature miRNA arms to the 5p (or 3p) human mature miRNA arms derived from the same orthologous precursor miRNA pair.
Identification of marker miRNAs
We used an elastic net regression model (implemented in the R package glmnet functionCitation19) to regress chronologic age on miRNA expression levels (log transformed transcripts/million [TPM] values). The alpha parameter of glmnet was set to 0.5 and the lambda value was set to 0.2 as suggested by Huan et al.Citation20 The miRNAs with non-zero coefficients were kept as candidate markers.
At the same time, we used a linear mixed-effects model to regress miRNA expression levels (log transformed TPM values) on chronologic age, adjusting for sex and family structure. The data analysis was performed utilizing the lmekin () R function (http://cran.r-project.org/web/packages/kinship/).Citation21 The miRNAs with Benjamini–Hochberg-corrected p values < 0.05 based on the linear mixed-effects model were also identified as candidate markers.
The candidate miRNAs identified by both methods were considered marker miRNAs.
Results
The overview of small-RNA sequences
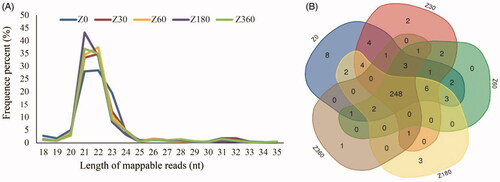
A total of 221.3 million raw reads were obtained from the peripheral blood of individual pigs drawn at the ages of 0, 30, 60, 180 and 360 days, using three biological replicates for each stage. The average raw read of each sample was 14.75 million (14.75 ± 1.46). After filtering low quality reads and adaptor sequences, we obtained 208.5 million high-quality reads (Table S2). Most of these reads were 21–23 nt in length (81.2 ± 5.18%, n = 15), with the highest proportion being 21 nt (35.24 ± 9.99%, n = 15), followed by 22 nt (33.97 ± 4.09%, n = 15) and 23 nt (12.0 ± 5.65%; ). We identified 295 known mature miRNAs corresponding to 287 precursors, 248 of which were expressed in all of the samples across all different developmental stages (). High Pearson’s correlations (average R2 = 0.96) between pairwise comparisons of samples indicated high similarity among samples ().
Figure 1. (A) Length distribution of high-quality miRNA reads at each postnatal developmental time point. (B) Venn diagram showing overlapping expression of individual miRNAs across the five developmental stages.
Global expression profile pattern of miRNAs across five developmental stages
The average miRNA expression levels of three biological replicates for each time point were applied to represent each stage. The 17 most highly expressed miRNAs across the five stages (), defined as miRNAs shared among the 20 most highly expressed miRNAs at each developmental stage, are listed in Table S3. These 17 miRNAs account for more than 80% of total miRNA expression and were over-represented in cellular metabolic processes, blood coagulation, and nervous and skeletal system development (Tables S4 and S5). This result indicated that these miRNAs are likely to play housekeeping roles in blood development and maintenance of basic cellular metabolism in blood.Citation22
Figure 2. (A) Upset plot of 17 microRNAs with the highest expression across the five developmental stages. (B) Heatmap of hierarchical clustering results based on pairwise Spearman’s correlations of miRNA expression levels between different stages. (C) Principal component (PC) analysis based on the miRNA expression profile across five developmental stages. (D) Over-represented temporal expression profiles with p < 0.05. The number in the top left (0, 21, 2, 48) of each box is the profile ID assigned by STEM. The number in the bottom left of each box (9 × 10−45, 9 × 10−18, 2 × 10−19, 2 × 10−4) is the adjusted p value. (E) Differentially expressed miRNAs of pairwise comparisons between time points. Graphs depict the log2-transformed fold-change in expression (x-axis) and minus-log10-transformed p values (y-axis).
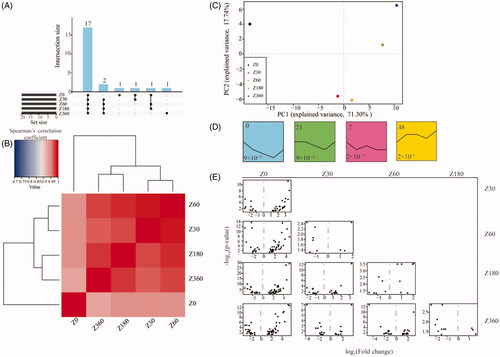
Hierarchical clustering based on Spearman’s correlations of miRNA expression levels between different stages showed that the stages could be separated into two age groups, 0 days and older, which suggested that there were significant differences between individuals at the neonatal and postnatal stages. The postnatal stage samples could be further segregated into two groups: weaning and weaned (Z30 and Z60), and puberty (Z180 and Z360; ). This result was consistent with our principal component analysis ().
STEM analysis was also used to group miRNAs into distinct time-series expression patterns, identifying four significant model profiles (). In model profile 0, the miRNA expression levels showed a steady decrease from 0 to 180 days, then reversed, showing a steady increase from 180 to 360 days. The miRNA expression levels in profile 48 exhibited an increase in the beginning, remained stable in the intermediate stages, then increased slightly in the later stages. Interestingly, the miRNAs in profile 0, the most over-represented profile, were mainly enriched in genes involved in cell proliferation, differentiation-related pathways, fatty acid biosynthesis and the T cell receptor signaling pathway (p < 0.05; Tables S6 and S7; ).
Differential expression and functional enrichment of miRNAs
Consistent with the observation that the global expression profile showed a clear separation between the neonatal stage and the four postnatal stages, there was a greater number of DE miRNAs between the neonatal and postnatal stages than between any single pair of postnatal stages ( and S8).
Table 1. Number of microRNAs differentially expressed between five postnatal developmental stages.
Given the time-course nature of the miRNA expression profile, we first identified genes that were DE between adjacent stages. Between the ages of 0 and 30 days, we identified 34 DE miRNAs enriched in genes involved in cellular lipid metabolic and protein modification processes, the innate immune response and immune-related signal pathways (p < 0.05; Tables S9 and S10; ). Nine miRNAs DE between the ages of 30 days and 60 days were identified, with over-representation in muscle cell differentiation, viral process and the B cell receptor signaling pathway and other immune-related signaling pathways (p < 0.05; Tables S11 and S12; ). Between the ages of 180 days and 360 days, 12 DE miRNAs were identified and found to be mainly enriched in blood coagulation, post-translational protein modification, platelet activation, and immune system response (p < 0.05; Tables S13 and S14; ). These results indicated that these DE miRNAs might play important roles in immune system development and metabolic regulation of postnatal growth.
Figure 3. Over-represented Gene Ontology (GO) terms (A) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (B) in the miRNAs that were differentially expressed among all of the pairwise comparisons between the neonatal (day 0) and four postnatal stages.
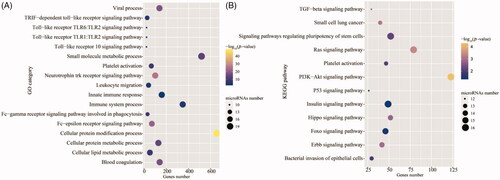
Figure 4. (A) Similarity among the 10 sets of differentially expressed (DE) miRNAs in pairwise comparisons between different stages. (B) Similarity of biologic processes enriched in the 10 between-stage comparisons. Over-represented temporal expression patterns of 3 miRNA subsets involved in the immune system (C), innate immune response (D), and toll-like receptor (tlr) signaling pathway (E), extracted from the ensemble of miRNAs that were DE in at least one between-stage comparison. The number in the top left of each box is the profile ID assigned by STEM. The number in the bottom left of each box is the adjusted p value. (F) Heatmap of hierarchical clustering results for 19 miRNAs exhibiting the steady decrease model profile (profile 9) for at least one of the three immune-related categories (C–E). ssc-miR: porcine miRNA; TPM: transcripts/million; TRIF: TIR-domain-containing adapter-inducing interferon-β.
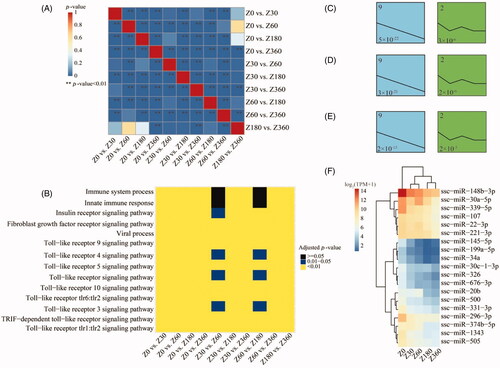
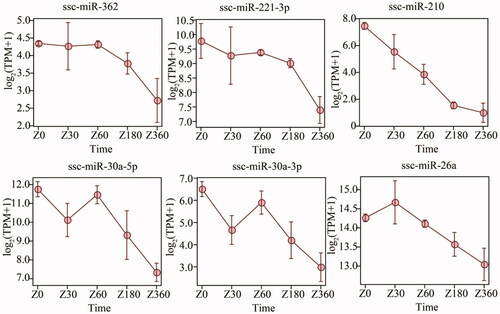
Figure 5. Temporal expression profiles for chronologic age-associated marker miRNAs (miRs). TPM: transcripts/million.
As the neonatal and postnatal stages showed the largest discrepancy, we identified 20 miRNAs that were common to the 4 sets of miRNAs that were DE between the ages of 0 days and each of the other four stages: that is, Z0 vs. Z30, Z0 vs. Z60, Z0 vs. Z180, Z0 vs. Z360; ; Tables S15 and S16). These DE miRNAs were enriched in blood coagulation, the innate immune response, leukocyte migration, immune system processes, cell proliferation-related signal pathways (ErbB, Hippo, Ras and p53), and immune-related pathways (Fc-epsilon receptor signaling and toll-like receptor [TLR] signaling pathways). The over-represented Fc-epsilon receptor signaling pathway, for example, plays a critical role in controlling the pathology of IgE-mediated allergies.Citation23 The enriched TLR signaling pathway plays a critical role in detecting microbial infection and triggering the host defense.Citation24 Because the period from birth to 4 weeks of age has been described as a period of immunodeficiency,Citation25 these DE miRNAs may be essential for the establishment and development of the peripheral blood-based immune system.
Immune-related miRNAs
Given that 20 DE miRNAs were common to the four pairwise comparisons of the neonatal and postnatal stages, we further compared the 10 sets of DE miRNAs for all of the pairwise comparisons between distinct stages. We observed that the DE miRNAs were similar for the majority of these comparisons (Chi-square test, p < 0.05; ). Interestingly, we noted recurrences of the following biologic processes enriched in genes that were DE among all of the between-stage comparisons: the innate immune response, immune system processes, the TLR signaling pathway, and viral process (). These DE miRNAs may indeed regulate similar biologic processes and signaling pathways.
To further explore the characteristics of the temporal miRNA expression profiles for these immune-related pathways, we extracted three miRNA subsets involved in the TLR signaling pathway, the innate immune response, or the immune system response, from the miRNA ensemble DE in at least one pairwise between-stage comparison (Tables S17–S19). STEM analysis of the expression profiles revealed that all three miRNA subsets were consistently over-represented in an expression pattern that steadily decreased over time ().
From the miRNAs exhibiting a pattern of steadily decreasing expression for at least one of the three immune-related categories, we extracted 19 miRNAs (), many of which were previously reported to be associated with physiologic and pathologic processes of immunity and inflammation. Among these is MiR-199a which was reported to be an important participant in immune disease multiple sclerosis (MS). And the miR-199a-3p level is associated with maturation of dendritic immune cells and secretion of inflammatory factors.Citation26,Citation27 Another, miR-145, is able to target the adaptor molecule TIRAP, which helps to connect TLR2- and TLR4-related myd88-dependent signaling.Citation28 The 5p arm of miR-30a can function as a regulator of glycolysis, which is important for regulating the growth of breast tumors.Citation29 Additionally, the overexpression of miR-30a impairs the anti-inflammatory effect of mesenchymal stem cells on macrophages.Citation30 In addition to inhibiting viral replication, miR-221 plays an critical role in regulating the processes of differentiated mast cell, such as degranulation and adhesion.Citation12,Citation31 In endothelial cells, overexpression of miR-22 promotes the synthesis of proinflammatory cytokines.Citation32
Chronologic age-associated marker miRNAs
We regressed the chronologic age on miRNA expression levels and vice versa using an elastic net regression model and a linear mixed-effects model. Six miRNAs that showed a significant association with chronologic age in both models were identified as marker miRNAs. Consistent with previous results, these marker miRNAs showed negative correlations with increasing age and were over-represented in immune-related biologic processes. ssc-miR-30a-5p, ssc-miR-30a-3p, ssc-miR-26a and ssc-miR-221 over-represented in TLR 10 signaling pathway, ssc-miR-30a-5p, ssc-miR-30a-3p, ssc-miR-26a and ssc-miR-362 also involved in Fc-epsilon receptor signaling pathway and viral process, leukocyte migration immune related biologic process (p < 0.05, ; Tables S20).
Discussion
In mammals, blood is both an informative and a dynamic type of tissue that contains a variety of RNAs.Citation33,Citation34 Gene expression profiles of blood can reflect the physio-pathologic status, growth stage and lifestyle of the host.Citation33,Citation35,Citation36 Blood contains multiple type of cells, including red blood cells (RBCs) and white blood cells (WBCs). WBCs can be further categorized into neutrophils, eosinophils, B lymphocytes, T lymphocytes and monocytes. Each of these cell types has a unique function. RBCs can transport oxygen and carbon dioxide, while neutrophils, eosinophils and basophils play important roles in inflammation.Citation37 B lymphocytes are one of the most important cell types involved in the immune response of mammals,Citation38 while T lymphocytes are responsible for cell-mediated immunity.Citation39 The immune system of mammals is not fully functional at birth, but rather undergoes age-dependent maturation.Citation40 The varied immune-related cell types found in peripheral blood, together with its easy accessibility and reflective relationship with physiologic status, make it an ideal investigative tissue with which to better understand this immune system maturation process.
In this study, we identified 295 known mature miRNAs from peripheral blood samples of 15 pigs across five different developmental stages. Our first observation was that the miRNAs were expressed in a stage-specific manner across different developmental stages. Hierarchical clustering of the samples revealed two groups: newborn piglets (0 days) and pigs of other developmental stage (30 days, 60 days, 180 days, 360 days). Pigs of other developmental stages were divided into two groups, weaning and weaned pigs (30 days and 60 days), and pigs in the later developmental stages of puberty (180 days and 360 days). This indicated that the miRNA expression levels were reflective of different developmental stages. Notably, miRNA expression in newborn piglets exhibited the largest discrepancies from the other four stages, which is consistent with previous reports that piglets from birth to 4 weeks of age suffer from immunodeficiency.Citation25
Second, STEM analyses showed that the temporal expression patterns of miRNAs were actually diverse, with the most over-represented profile demonstrating steady decreases over time (), which implied that different miRNAs might play distinct roles in regulating physiologic processes and participating in postnatal development.
Third, pathways for immune system processes, the innate immune response and TLR signaling were universally over-represented in all 10 sets of miRNAs that were DE in pairwise comparisons of distinct developmental stages, suggesting potentially important roles for miRNA in immune system development. Interestingly, DE miRNAs involved in these immune-related biologic processes showed enrichment in a pattern of steadily declining expression across the developmental stages. Because miRNAs are known to negatively regulate target coding genes by either inhibiting translation or inducing mRNA degradation, our results suggested that immune-related pathways regulated by these DE miRNAs might be steadily enhanced during development; thus, these miRNAs may contribute to age-dependent immune system maturation. This also accords with previous reports that the human immune system, particularly the innate immune system, continues to develop from the newborn stage into puberty.Citation41
Combining elastic net regression and linear mixed-effects models, we identified six age-associated marker porcine miRNAs (ssc-miR-362, ssc-miR-221-3p, ssc-miR-210, ssc-miR-30a-5p, ssc-miR-30a-3p and ssc-miR-26a). Interestingly, all six miRNAs showed negative correlation of expression level with age () and exhibited over-representation in immune-related pathways (TLR 10 signaling pathway, viral process, Fc-epsilon receptor signaling pathway, viral process, leukocyte migration) (p < 0.05). Specifically, overexpression of miR-30a in bacterial infected THP-1 cells inhibits the activation of TLR/MyD88.Citation42 In rat macrophages, it has been reported that miR-26a can negatively regulate the TLR 3 signaling pathway by targeting TLR 3 itself.Citation43 Another miRNA, miR-221, boost mast cell degranulation through PI3K/Akt signaling pathway and miR-221-5p impairs porcine epidemic virus replication by activating the NF-kB signaling pathway,Citation12,Citation31,Citation44 and miR-362 plays a key role in cell proliferation and tumor occurrence.Citation45,Citation46
In summary, our study provides an extensive resource for neonatal and postnatal stage-related miRNA expression in pig peripheral blood and may offer insight into immune system development in the pig. The subset of over-represented of immune-related miRNAs with steadily declining expression over time may serve as an indicator for the continuous development of the pig immune system from the newborn stage into puberty. This interpretation was further supported by the observation that age-associated marker miRNAs exhibited negative correlations with age and were over-represented in immune-related biologic processes.
Supplemental Material
Download MS Word (122.6 KB)Supplemental Material
Download MS Word (757 KB)Acknowledgments
We thank Michelle Kahmeyer-Gabbe, PhD, from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Disclosure statement
The authors declare that there are no conflicts of interest.
Data availability statement
All sequence data from this study was deposited in NCBI’s Gene Expression Omnibus series under the accession number GSE140780.
Additional information
Funding
References
- Butler JE, Sinkora M. The isolator piglet: a model for studying the development of adaptive immunity. Immunol Res. 2007;39(1–3):33–51.
- Butler J. Isolator and other neonatal piglet models in developmental immunology and identification of virulence factors. Anim Health Res Rev. 2009;10(1):35–52.
- Butler JE, Lager KM, Golde W, et al. Porcine reproductive and respiratory syndrome (PRRS): an immune dysregulatory pandemic. Immunol Res. 2014;59(1–3):81–108.
- Sun RQ, Cai RJ, Chen YQ, et al. Outbreak of porcine epidemic diarrhea in suckling piglets. Emerg Infect Dis. 2012;18(1):161–163.
- Shibata I, Tsuda T, Mori M, et al. Isolation of porcine epidemic diarrhea virus in porcine cell cultures and experimental infection of pigs of different ages. Veterinary Microbiology. 2000;72(3-4):173–182.
- Lee C. Porcine epidemic diarrhea virus: An emerging and re-emerging epizootic swine virus. Virol J. 2015;12:193.
- Lu L-F, Liston A. MicroRNA in the immune system, microRNA as an immune system. Immunology. 2009;127:291–298.
- Chaussabel D, Pascual V, Banchereau J. Assessing the human immune system through blood transcriptomics. BMC Biol. 2010;8:84.
- Seddiki N, Brezar V, Ruffin N, Lévy Y, Swaminathan S. Role of miR-155 in the regulation of lymphocyte immune function and disease. Immunology. 2014;142(1):32–38.
- Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc Natl Acad Sci USA. 2007;104(17):7080–7085.
- Liu Z, Xiao B, Tang B, et al. Up-regulated microRNA-146a negatively modulate Helicobacter pylori-induced inflammatory response in human gastric epithelial cells. Microbes Infect. 2010;12:854–863.
- Zheng H, Xu L, Liu Y, et al. MicroRNA-221-5p inhibits porcine epidemic diarrhea virus replication by targeting genomic viral RNA and activating the NF-κB pathway. IJMS. 2018;19(11):3381.
- Jia X, Bi Y, Li J, et al. Cellular microRNA miR-26a suppresses replication of porcine reproductive and respiratory syndrome virus by activating innate antiviral immunity. Sci Rep. 2015;5(1):10651.
- Ernst J, Bar-Joseph Z. STEM: a tool for the analysis of short time series gene expression data. BMC Bioinformatics. 2006;7(1):191.
- Ernst J, Nau GJ, Bar-Joseph Z. Clustering short time series gene expression data. Bioinformatics. 2005;21:i159–i68.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
- Vlachos IS, Zagganas K, Paraskevopoulou MD, et al. DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res. 2015;43:W460–W6.
- Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47(D1):D155–D62.
- Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1–22.
- Huan T, Chen G, Liu C, et al. Age-associated microRNA expression in human peripheral blood is associated with all-cause mortality and age-related traits. Aging Cell. 2018;17(1):e12687.
- Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62(5):1198–1211.
- Copley MR, Babovic S, Benz C, et al. The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat Cell Biol. 2013;15:916–925.
- Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat Rev Immunol. 2008;8(3):205–217.
- Lim KH, Staudt LM. Toll-like receptor signaling. Cold Spring Harb Perspect Biol. 2013;5(1):a011247–a011247.
- Layton HW, Simkins KL, Eggert RG. Efficacy of nithiamide in preventing and treating swine dysentery in experimentally inoculated pigs. Am J Vet Res. 1977;38:627–631.
- Ghadiri N, Emamnia N, Ganjalikhani-Hakemi M, et al. Analysis of the expression of mir-34a, mir-199a, mir-30c and mir-19a in peripheral blood CD4 + T lymphocytes of relapsing-remitting multiple sclerosis patients. Gene. 2018;659:109–117.
- Xiong A, Wang J, Mao XL, Jiang Y, Fan Y. MiR-199a-3p modulates the function of dendritic cells involved in transplantation tolerance by targeting CD86. Hla. 2019;94(6):493–503.
- Starczynowski DT, Kuchenbauer F, Argiropoulos B, et al. Identification of miR-145 and miR-146a as mediators of the 5q–syndrome phenotype. Nat Med. 2010;16(1):49–58.
- Li L, Kang L, Zhao W, et al. miR-30a-5p suppresses breast tumor growth and metastasis through inhibition of LDHA-mediated Warburg effect. Cancer Lett. 2017;400:89–98.
- Hu E, Ding L, Miao H, et al. MiR-30a attenuates immunosuppressive functions of IL-1β-elicited mesenchymal stem cells via targeting TAB3. FEBS Lett. 2015;589:3899–3907.
- Mayoral RJ, Deho L, Rusca N, et al. MiR-221 influences effector functions and actin cytoskeleton in mast cells. PLoS One. 2011;6(10):e26133.
- Gu W, Zhan H, Zhou XY, et al. MicroRNA-22 regulates inflammation and angiogenesis via targeting VE-cadherin. FEBS Lett. 2017;591:513–526.
- Mohr S, Liew C-C. The peripheral-blood transcriptome: new insights into disease and risk assessment. Trends Mol Med. 2007;13(10):422–432.
- Ogawa M. Differentiation and proliferation of hematopoietic stem cells. Blood. 1993;81:2844–2853.
- Liu H, Smith TPL, Nonneman DJ, Dekkers JCM, Tuggle CK. A high-quality annotated transcriptome of swine peripheral blood. BMC Genomics. 2017;18(1):479.
- Chaussabel D. Assessment of immune status using blood transcriptomics and potential implications for global health. Semin Immunol. 2015;27:58–66.
- Geering B, Stoeckle C, Conus S, Simon H-U. Living and dying for inflammation: neutrophils, eosinophils, basophils. Trends Immunol. 2013;34:398–409.
- Nothelfer K, Sansonetti PJ, Phalipon A. Pathogen manipulation of B cells: the best defence is a good offence. Nat Rev Microbiol. 2015;13:173–184.
- Raff M. T and B lymphocytes and immune responses. Nature. 1973;242(5392):19–23.
- Martin R, Nauta AJ, Ben Amor K, et al. Early life: gut microbiota and immune development in infancy. Benef Microbes. 2010;1(4):367–382.
- Georgountzou A, Papadopoulos NG. Postnatal innate immune development: from birth to adulthood. Front Immunol. 2017;8:957.
- Wu Y, Sun Q, Dai L. Immune regulation of miR-30 on the Mycobacterium tuberculosis-induced TLR/MyD88 signaling pathway in THP-1 cells. Exp Ther Med. 2017;14(4):3299–3303.
- Jiang C, Zhu W, Xu J, et al. MicroRNA-26a negatively regulates toll-like receptor 3 expression of rat macrophages and ameliorates pristane induced arthritis in rats. Arthritis Res Ther. 2014;16(1):R9.
- Xu H, Gu L-N, Yang Q-Y, Zhao D-Y, Liu F. MiR-221 promotes IgE-mediated activation of mast cells degranulation by PI3K/Akt/PLCγ/Ca2+ pathway. J Bioenerg Biomembr. 2016;48(3):293–299.
- Luo D, Zhang Z, Zhang Z, et al. Aberrant expression of miR-362 promotes lung cancer metastasis through downregulation of Sema3A. J Immunol Res. 2018;2018:1687097.
- Wu F, Yin C, Qi J, et al. miR-362-5p promotes cell proliferation and cell cycle progression by targeting GAS7 in acute myeloid leukemia. Hum Cell. 2020;33(2):405–415.