
Abstract
Introduction of hydrogen bond functionality into metal-organic frameworks can enhance guest binding and activation, but a combination of linker flexibility and interligand hydrogen bonding often results in the generation of unwanted structures where the functionality is masked. Herein, we describe the self-assembly of three materials, where Cd2+, Ca2+, and Zn2+ are linked by N,Nʹ-bis(4-carboxyphenyl)urea, and examine the effect of the urea units on structure formation, the generation of unusual secondary building units, structural flexibility, and guest binding. The flexibility of the Zn MOF is probed through single-crystal to single-crystal transformations upon exchange of DMF guests for CS2, showing that the lability of the [Zn4O(RCO2)6] cluster towards solvation enables the urea linkers to adopt distorted conformations as the MOF breathes, even facilitating rotation from the trans/trans to the trans/cis conformation without compromising the overall topology. The results have significant implications in the mechanistic understanding of the hydrolytic stability of MOFs, and in preparing heterogeneous organocatalysts.
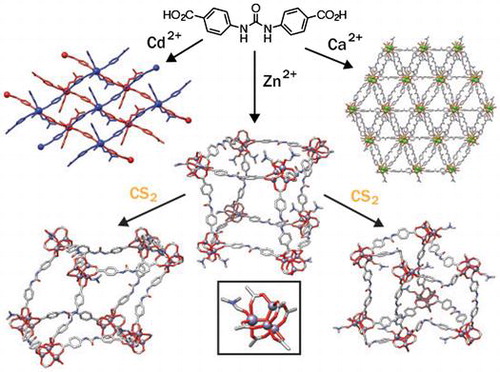
Introduction
Metal-organic frameworks (MOFs) are network materials comprising metal ion or cluster secondary building units (SBUs) linked by organic linkers into multidimensional structures that often exhibit considerable porosity (Citation 1–3 ). Many examples contain arrays of chemically addressable pores, with functionalised derivatives acting as ‘crystalline molecular flasks’ (Citation 4, 5 ) and finding application in guest binding and activation for heterogeneous catalysis (Citation 6–10 ). For example, grafting hydrogen bond donor functionality, such as amides (Citation 11–20 ), ureas (Citation 21–32 ), thioureas (Citation 33 ), and squaramides (Citation 34–36 ), (Figure (a)) onto the organic scaffold of the MOF can enhance properties including carbon dioxide capture (Citation 13, 15, 17, 18 ), anion binding (Citation 20, 21 ), sensing (Citation 28 ), and organocatalytic activity (Citation 6 ). Urea-functionalised MOFs in particular have been found to catalyse Friedel-Crafts reactions between pyrroles and nitroalkenes (Citation 22, 24, 27, 29, 31 ), Henry reactions between aldehydes and nitromethane (Citation 23 ), and the methanolysis of epoxides (Citation 25 ).
Figure 1. (Colour online) (a) H-bond donor groups incorporated into MOFs. (b) Structure-directing H-bonding between urea and carboxylate units of different nets in an interpenetrated Zn MOF (redrawn from CCDC deposition 1011797). (c) Chemical structure of the ligand N,Nʹ-bis(4-carboxyphenyl)urea (LH 2 ) used in this study. (d) Structures of the three different urea conformations.
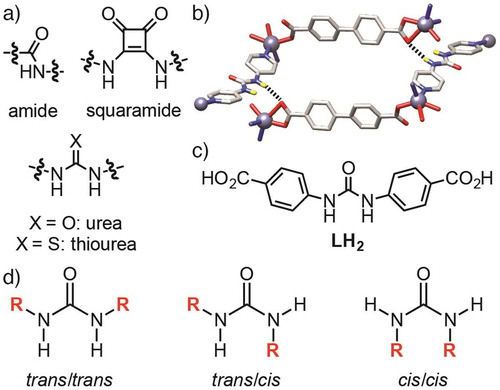
Direct synthesis of urea-containing MOFs, by introduction of urea moieties into organic linkers to target specific topologies in an isoreticular synthetic approach, is complicated by the possibility of structure-directing hydrogen bonding between the linkers inducing unexpected or unwanted MOF structures. For example, we have previously reported that the use of N,Nʹ-bis(4-pyridyl)urea instead of 4,4ʹ-bipyridine in solvothermal syntheses with Zn2+ sources and dicarboxylic acids results in MOFs with interpenetrated diamondoid topology, rather than the expected pillared primitive cubic topology, as a consequence of hydrogen bonding between the urea groups of the pillar and the carboxylate groups of the ligand directing structure formation (Figure (b)) (Citation 37 ). The formation of hydrogen bonds between different parts of the MOF structure could mask potential catalytic sites, and so an understanding of how it can be avoided when using ligands such as N,Nʹ-bis(4-carboxyphenyl)urea (LH 2 , Figure (c)) is essential. Additionally, hydrogen bonding moieties often introduce structural flexibility; urea units have rotational freedom around the C–N–C moieties and can adopt (Figure (d)) three different conformations – trans/trans, trans/cis, cis/cis – with the trans/trans conformation (also known as the syn, syn conformation) expected to be optimal for organocatalysis and guest binding (Citation 38, 39 ). Herein, we describe the direct self-assembly and solid-state structures of three coordination polymers of LH 2 and different metal cations, Cd2+, Ca2+ and Zn2+, discussing the effect of urea incorporation on structure, formation of novel inorganic SBUs, guest binding, and flexibility. We probe the guest binding properties of the Zn MOF with CS2 as a mimic for CO2, and show that incorporation of the guest induces single-crystal to single-crystal (SCSC) transformations with notable changes in both linker conformation and SBU coordination chemistry. These results have significant implications in the understanding of activation, guest binding and hydrolysis of H-bond functionalised MOFs.
Experimental
General
All chemicals and solvents were purchased from Alfa Aesar, Fisher Scientific, VWR, and Sigma Aldrich, and used as received.
Crystallography
Single Crystal X-ray diffraction data for [Cd(L)(DMF)3]n and [Ca5(L)5(DMF)3(H2O)2]n were collected using a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum rotating anode generator with VHF Varimax optics (70 μm focus) equipped with an Oxford Cryosystems cryostream device (EPSRC UK National Crystallography Service) (Citation 40 ). Data were collected using CrystalClear-SM Expert 3.1 b27 (Citation 41 ) and processed with CrysAlisPro 1.171.38.43 (Citation 42 ). Single crystal data for both solvates of [Zn4O(L)3(DMF)2]n as well as their daughter products isolated during soaking in CS2 were collected using a Bruker D8 Venture goniometer with a Bruker PHOTON II detector and dual ImuS 3.0 microfocus sources (Cu and Mo Kα) equipped with an Oxford Cryosystems n-Helix device. Mo Kα radiation was used for all data collections except [Zn4O(L)3(DMF)(H2O)]n where Cu Kα was used (University of Glasgow). Data were collected using APEX3 Ver. 2016.9-0 (Citation 43 ) and processed with SAINT V8.37A (Citation 44 ).
The structures were solved using ShelxT (Citation 45 ) and refined against F 2 using Shelx2015 (Citation 46 ) within Olex2 (Citation 47 ). [Cd(L)DMF3]n was treated as a two component twin related by a 2-fold rotation about the 100 direction. The twin component fractions refined to 0.421(6)/0.579(6) giving significant improvement, however, probably due to some unaccounted for twinning, only the Cd atom was refined with anisotropic atomic displacement parameters (adps). For this structure SQUEEZE (Citation 48 ) was only used to calculate the solvent accessible volume (32 Å3) however for all other structures reported herein SQUEEZE was used to calculate and account for the electron density within the solvent accessible void; details are given in Table . Disorder was present and modelled as two 0.5 occupied sites in one linker for both [Ca5(L)5(DMF)3(H2O)2]n and [Zn4(L)3(DMF)(H2O)]n. Distance restraints were used in the case of the disordered fragments and for the solvent (DMF and CS2) geometry. For the samples treated with CS2 all the structures showed residual electron density on the Zn sites possibly due to unaccounted for twinning however treatment as a two component crystal was not satisfactory and was not used.
Table 1. Crystallographic data comparison.
Synthesis
LH 2 was synthesised by a modified literature procedure (Citation 49 ).
[Cd(L)(DMF) 3 ] n . Cadmium nitrate tetrahydrate (0.010 g, 0.032 mmol), LH 2 (0.009 g, 0.030 mmol) and N,N-dimethylformamide (DMF, 5 ml) were added to a 25 ml Pyrex reagent bottle and sonicated. The resulting solution was placed in the oven at 100 °C for 48 h. The bottle was removed from the oven after this period and allowed to cool to room temperature. The crystals were left to stand in their mother solution.
Crystal data for [Cd(L)(DMF) 3 ] n . C24H31CdN5O8, M r = 629.94, crystal dimensions 0.07 × 0.05 × 0.01 mm, Monoclinic, a = 9.1728 (7) Å, b = 15.2062 (14) Å, c = 19.768 (2) Å, V = 2749.5 (5) Å3, T = 100 K, space group P21 /n (no. 14), Z = 4, 18,150 measured reflections, 4,767 unique (R int = 0.147). Data were not merged as from a two component twin and all 18,510 reflections were used in all calculations. The final R 1 = 0.161 for 11,215 (of 18510) observed data R[F 2 > 2σ(F 2)] and wR(F 2) = 0.389 (all data). Crystal structure data are available from the CCDC, deposition number 1558143.
[Ca 5 (L) 5 (DMF) 3 (H 2 O) 2 ] n . Calcium nitrate tetrahydrate (0.008 g, 0.033 mmol), LH 2 (0.010 g, 0.033 mmol) and DMF (10 ml) were added to a 50 ml Pyrex reagent bottle and sonicated. The resulting solution was placed in the oven at 100 °C for 48 h. The bottle was removed from the oven after this period and allowed to cool to room temperature. The crystals were left to stand in their mother solution.
Crystal Data for [Ca 5 (L) 5 (DMF) 3 (H 2 O) 2 ] n . C84H68Ca5N13O30·6.75(C3H7NO)·3.5(H2O), M r = 2503.42, crystal dimensions 0.22 × 0.06 × 0.04 mm, Triclinic, a = 16.0056 (4) Å, b = 18.4778 (6) Å, c = 31.0206 (4) Å, V = 8431.9 (4) Å3, T = 100 K, space group P-1 (no. 2), Z = 2, 91,045 measured reflections, 29,730 unique (R int = 0.083) which were used in all calculations. The final R 1 = 0.109 for 19,792 observed data R[F 2 > 2σ(F 2)] and wR(F 2) = 0.347 (all data). Approximately 30% of the cell volume is not occupied by the framework and contains diffuse and disordered solvent molecules. This electron density was accounted for using SQUEEZE within PLATON (Citation 48 ) which calculated a solvent accessible volume of 2565 Å3 containing 681 electrons (the equivalent of ~17 molecules of DMF) per unit cell. Crystal structure data are available from the CCDC, deposition number 1558144.
[Zn 4 O(L) 3 (DMF) 2 ] n -I. Zinc nitrate hexahydrate (0.050 g, 0.168 mmol), LH 2 (0.050 g, 0.167 mmol) and DMF (20 ml) were added to a 100 ml reagent bottle and sonicated. The resulting solution was placed in the oven at 90 °C for 24 h. The bottle was removed from the oven after this period and allowed to cool to room temperature. The crystals were left to stand in their mother solution. Crystals of [Zn4(L)3(DMF)2]n-II were also isolated from this synthesis in the same container. As the two solvates are identical in connectivity and topology they were not separated, and referred to as [Zn4(L)3(DMF)2]n.
Crystal Data for [Zn 4 O(L) 3 (DMF) 2 ] n -I. C51H44N8O18Zn4·6(C3H7NO), M r = 1756.99, crystal dimensions 0.16 × 0.12 × 0.07 mm, Monoclinic, a = 16.0084 (5) Å, b = 30.1963 (14) Å, c = 20.4622 (8) Å, V = 9778.3 (7) Å3, T = 100 K, space group P21 /c (no. 14), Z = 4, 55,552 measured reflections, 24,084 unique (R int = 0.052) which were used in all calculations. The final R 1 = 0.087 for 18,205 observed data R[F 2 > 2σ(F 2)] and wR(F 2) = 0.243 (all data). Approximately 10% of the cell volume is not occupied by the framework and contains diffuse and disordered solvent molecules. This electron density was accounted for using SQUEEZE within PLATON (Citation 48 ) which calculated a solvent accessible volume of 956 Å3 containing 154 electrons (the equivalent of ~3.85 molecules of DMF) per unit cell. Crystal structure data are available from the CCDC, deposition number 1558145.
Crystal Data for [Zn 4 O(L) 3 (DMF) 2 ] n -II. C51H44N8O18Zn4·3(C3H7NO), M r = 1537.71, crystal dimensions 0.26 × 0.24 × 0.17 mm, Monoclinic, a = 15.7982 (10) Å, b = 30.2971 (17) Å, c = 20.4468 (14) Å, V = 9644.8 (11) Å3, T = 100 K, space group P21 /c (no. 14), Z = 4, 123,520 measured reflections, 23,849 unique (R int = 0.102) which were used in all calculations. The final R 1 = 0.068 for 13,455 observed data R[F 2 > 2σ(F 2)] and wR(F 2) = 0.236 (all data). Approximately 33% of the cell volume is not occupied by the framework and contains diffuse and disordered solvent molecules. This electron density was accounted for using SQUEEZE within PLATON (Citation 48 ) which calculated a solvent accessible volume of 3164 Å3 containing 905 electrons (the equivalent of ~22.6 molecules of DMF) per unit cell. Crystal structure data are available from the CCDC, deposition number 1558146.
A small amount of [Zn4(L)3(DMF)2]n was removed from its mother solution by pipette (ca. 2 ml) and added to a scintillation vial containing CS2 (ca. 5 ml). The CS2 was exchanged for fresh CS2 multiple times to remove as much DMF as possible. The CS2 was replenished daily and single crystal X-ray diffraction data of daughter products, namely [Zn4O(L)3(DMF)(H2O)]n, [Zn4O(L)3]n and [Zn4O(L)3(DMF)]n were collected after 4, 19, and 25 days, in turn.
Crystal Data for [Zn 4 O(L) 3 (DMF)(H 2 O)] n . C48H39N7O18Zn4·0.5(H2O)·5(C3H7NO), M r = 1637.83, crystal dimensions 0.14 x 0.10 x 0.05 mm, Monoclinic, a = 18.3487 (7) Å, b = 29.8698 (11) Å, c = 16.0422 (7) Å, V = 8741.1 (6) Å3, T = 100 K, space group P21 /c (no. 14), Z = 4, 64,583 measured reflections, 17,834 unique (R int = 0.083) which were used in all calculations. The final R 1 = 0.126 for 12,400 observed data R[F 2 > 2σ(F 2)] and wR(F 2) = 0.390 (all data). Approximately 17% of the cell volume is not occupied by the framework and contains diffuse and disordered solvent molecules. This electron density was accounted for using SQUEEZE within PLATON (Citation 48 ) which calculated a solvent accessible volume of 1498 Å3 containing 241 electrons (the equivalent of ~6 molecules of DMF) per unit cell. Crystal structure data are available from the CCDC, deposition number 1558147.
Crystal Data for [Zn 4 O(L) 3 ] n . C45H30N6O16Zn4·3(C3H7NO)·9(CS2), M r = 2076.69, crystal dimensions 0.23 × 0.08 × 0.08 mm, Monoclinic, a = 18.6280 (12) Å, b = 26.2694 (18) Å, c = 19.2096 (13) Å, V = 9308.0 (11) Å3, T = 100 K, space group P21 /c (no. 14), Z = 4, 74,700 measured reflections, 17,012 unique (R int = 0.057) which were used in all calculations. The final R 1 = 0.116 for 12,061 observed data R[F 2 > 2σ(F 2)] and wR(F 2) = 0.385 (all data). Approximately 10% of the cell volume is not occupied by the framework and contains diffuse and disordered solvent molecules. This electron density was accounted for using SQUEEZE within PLATON (Citation 48 ) which calculated a solvent accessible volume of 968 Å3 containing 92 electrons (the equivalent of ~2.4 molecules of CS2) per unit cell. Crystal structure data are available from the CCDC, deposition number 1558148.
Crystal Data for [Zn 4 O(L) 3 (DMF)] n . C48H37N7O17Zn4·5.5(CS2)·C3H7NO, M r = 1737.13, crystal dimensions 0.12 × 0.11 × 0.10 mm, Monoclinic, a = 16.5296 (14) Å, b = 27.609 (3) Å, c = 19.6058 (18) Å, V = 8916.1 (14) Å3, T = 100 K, space group P21 /c (no. 14), Z = 4, 84,731 measured reflections, 16,308 unique (R int = 0.089) which were used in all calculations. The final R 1 = 0.116 for 10,175 observed data R[F 2 > 2σ(F 2)] and wR(F 2) = 0.390 (all data). Approximately 23% of the cell volume is not occupied by the framework and contains diffuse and disordered solvent molecules. This electron density was accounted for using SQUEEZE within PLATON (Citation 48 ) which calculated a solvent accessible volume of 2070 Å3 containing 792 electrons (the equivalent of ~20.8 molecules of CS2) per unit cell. Crystal structure data are available from the CCDC, deposition number 1558149.
All crystallographic data are summarised and compared in Table . CCDC 1558143–1558149 contain the supplementary crystallographic data. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre; see https://www.ccdc.cam.ac.uk/.
Results and discussion
The urea-dicarboxylate ligand, LH 2 , was prepared on the gram scale according to a modification of a literature procedure in two steps from t-butyl 4-aminobenzoate and carbonyldiimidazole (Citation 49 ). Attempts were subsequently made to prepare MOFs containing the ligand with free urea units to examine guest binding. Solvothermal synthesis with Cd(NO3)2·4H2O in DMF at 100 °C for 48 h resulted in the isolation of the one-dimensional coordination polymer [Cd(L)(DMF)3]n. The material consists of chains of L 2– molecules connected by seven-coordinate Cd2+ cations with distorted pentagonal bipyramidal geometry (Figure (a)). The Cd2+ centres coordinate to both oxygen atoms of the carboxylate units of two molecules of L 2– , linking them in a trans manner, with three DMF molecules occupying the remainder of the coordination sphere. The 1D chains that result have the urea N-H units projecting only in one direction for any given chain, and these form bifurcated H-bond interactions (Figure (b)) with the carboxylate units of the two L 2– molecules around the Cd2+ SBUs of adjacent chains (N1···O5 = 2.784, N1···O3 = 3.232 Å; N2···O3 = 2.785, N2···O5 = 3.431 Å). The linear packing arrangement of the chains results in 2D H-bonded grids, with all urea units involved in hydrogen bonding (Figure (c)). The grids then stack upon each other with no significant intermolecular interactions.
Figure 2. (Colour online) The solid state structure of [Cd(L)DMF3]n. (a) One dimensional chains of Cd2+ cations linked by molecules of L 2– . (b) Interligand hydrogen bonding between the urea units of molecules of L 2– from one chain and carboxylate units of molecules of L 2– from an adjacent chain. (c) Assembly of the one dimensional polymers of [Cd(L)(DMF)3]n into an infinite two dimensional grid structure, with chains of individual sheets coloured red and blue. H atoms other than the N-H units removed for clarity in part (b).
![Figure 2. (Colour online) The solid state structure of [Cd(L)DMF3]n. (a) One dimensional chains of Cd2+ cations linked by molecules of L 2– . (b) Interligand hydrogen bonding between the urea units of molecules of L 2– from one chain and carboxylate units of molecules of L 2– from an adjacent chain. (c) Assembly of the one dimensional polymers of [Cd(L)(DMF)3]n into an infinite two dimensional grid structure, with chains of individual sheets coloured red and blue. H atoms other than the N-H units removed for clarity in part (b).](/cms/asset/a6bc1c49-1f9e-4c5b-8f16-468c74e64c54/gsch_a_1370095_f0002_oc.jpg)
The seven coordinate SBU has only been observed in one other Cd2+ coordination polymer – a related material with tetrabromoterephthalate linkers (Citation 50 ) – but derivatives where the DMF ligands are replaced by water to form coordination polymers are also known (Citation 51, 52 ). The urea units of L 2– clearly direct the formation of this structure, and would be unavailable for guest binding.
When LH 2 is combined with Ca(NO3)2·4H2O in DMF and heated to 100 °C for 48 h, a very small quantity of crystals of [Ca5(L)5(DMF)3(H2O)2]n results. The structure contains infinite chains of calcium cations linked by carboxylate units of L 2– . There are five crystallographically independent Ca2+ cations in the chains, but each has the same overall seven-coordinate, distorted pentagonal bipyramidal geometry (Figure (a)).
Figure 3. (Colour online) (a) Schematic of the coordination sphere of the Ca2+ ions in [Ca5(L)5(DMF)3(H2O)2]n. (b) The coordination environments of the five crystallographically independent Ca2+ ions in the crystal structure of [Ca5(L)5(DMF)3(H2O)2]n. (c) Extended structure of [Ca5(L)5(DMF)3(H2O)2]n viewed down the crystallographic a axis, showing triangular channels. (d) Bifurcated hydrogen bonding of a DMF molecule by the urea unit of one of the linkers, also showing the two coordination motifs to the Ca2+ cations in the structure. (e) Alternative view down the crystallographic b axis, showing further small pores. Disorder, non-coordinated solvents and hydrogen atoms removed for clarity.
![Figure 3. (Colour online) (a) Schematic of the coordination sphere of the Ca2+ ions in [Ca5(L)5(DMF)3(H2O)2]n. (b) The coordination environments of the five crystallographically independent Ca2+ ions in the crystal structure of [Ca5(L)5(DMF)3(H2O)2]n. (c) Extended structure of [Ca5(L)5(DMF)3(H2O)2]n viewed down the crystallographic a axis, showing triangular channels. (d) Bifurcated hydrogen bonding of a DMF molecule by the urea unit of one of the linkers, also showing the two coordination motifs to the Ca2+ cations in the structure. (e) Alternative view down the crystallographic b axis, showing further small pores. Disorder, non-coordinated solvents and hydrogen atoms removed for clarity.](/cms/asset/4e6f2033-4c25-4828-8ac8-376c2a5885eb/gsch_a_1370095_f0003_oc.jpg)
One carboxylate unit of L 2– chelates with both its oxygen atoms to the Ca2+ cation, with each oxygen coordinating to an adjacent calcium centre to overall bridge three Ca2+ cations in a (η 2:η 2:μ3) fashion. Two further carboxylate units of L 2– molecules bridge from the Ca2+ cation to adjacent Ca2+ centres above and below, both in a (η 1:η 1:μ2) motif. Each Ca2+ therefore coordinates to six carboxylate oxygen atoms from five different ligands, with a final DMF or water molecule making up the coordination sphere (Figure (b)). Each molecule of L 2– links the infinite 1D chains of Ca2+ ions through a (η 2:η 2:μ3) motif at one carboxylate and a (η 1:η 1:μ2) motif at the other. The chains run down the crystallographic a axis, and are linked into an approximately hexagonal array by molecules of L 2– .
There are five crystallographically independent linker molecules, all of which experience some distortion from an idealised planar structure. Small triangular pores run down the crystallographic a axis (Figure (c)) and these spaces are filled in the crystal structure with a large number of water and DMF solvent molecules, with two of the five independent urea units binding water guests through a bifurcated H-bonding motif (N1C···O5 W = 3.381, N2C···O5 W = 2.940 Å; N1D···O4 W = 2.827, N2D···O4 W = 3.005 Å) and a further two urea units binding DMF molecules (N1···O61S = 2.832, N2···O61S = 2.801 Å; N1A···O56S = 3.377, N2A···O56S = 2.809 Å) in a similar manner (Figure (d)). There are also small voids between the chains perpendicular to the channels (Figure (e)). The 1D chains of Ca2+ cations units in [Ca5(L)5(DMF)3(H2O)2]n enforce a topology in which the urea moieties are not involved in any inter-ligand hydrogen bonding. Unfortunately we were unable to find suitable synthetic conditions to access any more than a few crystals per reaction, and so alternative MOFs were sought for study.
Solvothermal reaction of LH 2 with Zn(NO3)2·6H2O in DMF for 24 h at 90 °C yielded block-shaped crystals of [Zn4O(L)3(DMF)2]n-I, which has the well-known IRMOF topology first reported by Yaghi in 1999 for MOF-5 (also known as IRMOF-1), [Zn4O(bdc)3]n where bdc = 1,4-benzenedicarboxylate (Citation 53 ). The material has a slightly different SBU than the parent IRMOF structure; instead of four tetrahedral Zn2+ cations linked by a μ4-O2– and six carboxylates, there are three tetrahedral Zn2+ centres and one octahedral Zn2+ cation with two additional coordinated DMF ligands (Figure (a)). This solvated SBU is rarely seen in IRMOF structures, but is known to occur when IRMOFs are prepared from linkers that are flexible (Citation 54, 55 ) or deviate from ideal linear geometry (Citation 56, 57 ). Whilst the L 2– linkers lie along linear edge positions connecting adjacent Zn4O SBUs in the primitive cubic topology, the carboxyphenyl groups are disposed at approximately 145° rather than 180°, and the linkers also bow in and out of the plane, indicating significant flexibility (Citation 58 ). Indeed, [Zn4O(L)3(DMF)2]n-I is structurally similar to NJU-Bai2, a Zn2+ IRMOF prepared from an analogous ligand with a central amido group rather than a urea moiety (Citation 59 ). [Zn4O(L)3(DMF)2]n-I is triply interpenetrated, with three identical nets nested within one another (Figure (b)), but surprisingly there are no net-net hydrogen bonding interactions between the urea and carboxylate functionalities. Instead, in the crystal structure each of the three crystallographically independent urea units binds a DMF molecule through bifurcated hydrogen bonding to the formamide oxygens (N1···O40T = 2.696, N2···O40T = 2.942 Å; N1A···O11S = 2.848, N2A···O11S = 2.863 Å; N1B···O21S = 2.967, N2B···O21S = 2.850 Å).
Figure 4. (Colour online) The solid-state structure of [Zn4O(L)3(DMF)2]n-I. (a) A portion showing the IRMOF topology, with the solvated SBU inset. (b) The three interpenetrated nets are shown in red, green and blue. Non-coordinated solvent, H atoms and disorder removed for clarity. (c) Microscope image of the two habits (large blocks and agglomerated plates) observed in solvothermal syntheses of [Zn4O(L)3(DMF)2]n, corresponding to the two solvates which crystallise in the same vessel.
![Figure 4. (Colour online) The solid-state structure of [Zn4O(L)3(DMF)2]n-I. (a) A portion showing the IRMOF topology, with the solvated SBU inset. (b) The three interpenetrated nets are shown in red, green and blue. Non-coordinated solvent, H atoms and disorder removed for clarity. (c) Microscope image of the two habits (large blocks and agglomerated plates) observed in solvothermal syntheses of [Zn4O(L)3(DMF)2]n, corresponding to the two solvates which crystallise in the same vessel.](/cms/asset/d25e5a91-dcc6-49b9-abed-be606c2832ab/gsch_a_1370095_f0004_oc.jpg)
Interestingly, a second set of crystals was observed after the solvothermal synthesis with different morphology – agglomerates of plates (Figure (c)) – which single crystal X-ray diffraction revealed to be a closely related material, [Zn4O(L)3(DMF)2]n-II, with identical connectivity and topology to [Zn4O(L)3(DMF)2]n-I but a slightly different unit cell, presumably due to differing levels of solvation. Isolation of this additional phase is likely a consequence of the flexibility of the MOF allowing for crystallisation of closely related solvates from one synthesis. As the topology and structure of the two are identical, and because mechanical manipulation would be the only possible method of separation, we refer to the combined material as simply [Zn4O(L)3(DMF)2]n in the remainder of the study.
We first reported the isolation of this material in our previous study of urea incorporation into MOFs (Citation 37 ) but did not fully describe the structure. During the preparation of this manuscript, a report detailing the synthesis of [Zn4O(L)3(DMF)2]n and its catalytic activity in the Friedel-Crafts reaction between indole and β-nitrostyrene was published (Citation 29 ). The fact that the urea groups are not involved in ‘host-host’ hydrogen bonding facilitates this reported catalytic activity once the bound DMF molecules are removed, further highlighting the importance of limiting these interactions by topological control during the design of organocatalytic MOFs.
As part of our attempts to understand the availability of the urea groups for guest binding within the pores of this MOF, we attempted to replace the DMF solvents of crystallisation with CS2 by soaking crystals of the MOF in this liquid CO2 mimic. This approach has been successfully used to examine guest binding in coordination cages (Citation 60 ), but reported structures of MOFs with bound CS2 remain relatively rare (Citation 61–67 ). As with the recently published study (Citation 29 ), [Zn4O(L)3(DMF)2]n was found to be relatively unstable to solvent removal, but CS2 is a volatile nonpolar solvent that may allow for efficient activation, and samples maintained crystallinity on solvent exchange from DMF to CS2. Single crystal X-ray diffraction analysis on a sample that had been soaked in CS2 at room temperature for 4 days revealed that rather than exchanging bound DMF guests for CS2, one of the DMF ligands coordinated to the octahedral Zn2+ cation in [Zn4O(L)3(DMF)2]n (Figure (a)) was replaced with water in a SCSC transformation to form [Zn4O(L)3(DMF)(H2O)]n (Figure (b)). It has been shown previously that DMF can dynamically bind to the metal cluster in the archetypal material MOF-5 (Citation 68 ), and these results clearly demonstrate that, even in MOFs where the DMF interacts with the SBU sufficiently to be located crystallographically, it can still be exchanged under mild conditions for alternative ligands. This partially hydrated cluster could also be considered as an intermediate species in the hydrolysis of the MOF, and indeed a model for all Zn MOFs that contain the basic zinc acetate SBU, which are known to be particularly susceptible to hydrolysis (Citation 69 ). Whilst the topology of the MOF remains unchanged, the flexibility is apparent when comparing [Zn4O(L)3(DMF)(H2O)]n to the parent structure. The ability of the ligand to distort, combined with the labile coordination chemistry of the SBU, allows the MOF to deviate from an approximately cubic arrangement (Figure (a)) to a flatter structure reminiscent of a rhombohedron (Figure (b)).
Figure 5. (Colour online) Comparison of the solid-state structures of the Zn MOFs of L 2− , showing the topology and the SBU. (a) The parent MOF, [Zn4O(L)3(DMF)2]n, from which (b) [Zn4O(L)3(DMF)(H2O)]n, (c) [Zn4O(L)3]n, and (d) [Zn4O(L)3(DMF)]n were prepared by single-crystal to single-crystal transformations. Non-coordinated solvent, H atoms and disorder removed for clarity.
![Figure 5. (Colour online) Comparison of the solid-state structures of the Zn MOFs of L 2− , showing the topology and the SBU. (a) The parent MOF, [Zn4O(L)3(DMF)2]n, from which (b) [Zn4O(L)3(DMF)(H2O)]n, (c) [Zn4O(L)3]n, and (d) [Zn4O(L)3(DMF)]n were prepared by single-crystal to single-crystal transformations. Non-coordinated solvent, H atoms and disorder removed for clarity.](/cms/asset/7e66fe41-d100-4937-943b-2b6c7f5b81a9/gsch_a_1370095_f0005_oc.jpg)
Leaving the same batch of crystals in CS2 for a further 15 days, with daily replenishment of the CS2, resulted in a further SCSC transformation to [Zn4O(L)3]n, where all coordinated solvents had been removed and the SBU has the conventional Zn4O(RCO2)6 composition (Figure (c)). Additionally, guest exchange occurred, with significant quantities of CS2 now located and ordered within the pores of the MOF, replacing weakly-bound DMF molecules. Tellingly, the only DMF that remains within the pores is hydrogen bonded to the urea units (N1···O6D = 2.893, N2···O6D = 2.903 Å; N1A···O1D = 2.860, N2A···O1D = 2.843 Å; N1B···O11D = 2.863, N2B···O11D = 2.847 Å), indicating the strength of the interaction, with the CS2 located in the pores. While CS2 may be a good geometric mimic of CO2, it is a weaker hydrogen bond acceptor, likely due to it having an opposite quadrupole moment and thus not being electronically analogous (Citation 60 ), and it is certainly a weaker hydrogen bond acceptor than DMF.
The structure still has a rhombohedral-like arrangement, but seems visually less distorted than [Zn4O(L)3(DMF)(H2O)]n. To attempt to quantify the flexibility-induced distortion, the angles between adjacent SBUs were measured for each MOF, taking the bridging μ4-O2– units of the Zn4O SBUs as their centres, and collated in Table . For a perfect cubic structure, twelve angles of 90° would be expected, but the non-linear geometry of L 2− , coupled with its ability to bow out of the plane, means a range of angles are observed experimentally. The standard deviation was calculated as a metric to describe the magnitude of distortion, with larger standard deviations indicating more significant distortion from the idealised cubic arrangement. The two solvates of [Zn4O(L)3(DMF)2]n have similar values, while [Zn4O(L)3(DMF)(H2O)]n has the largest standard deviation, with angles ranging from ~60° to 120°, which is obvious from inspection of the structure (Figure (b)). [Zn4O(L)3]n lies between the two extremes.
Table 2. Angles measured between central μ4-O2– ligands of the Zn4O SBUs in the Zn MOFs of L 2– . All angles would be 90° in an ideal cubic structure; the largest standard deviation indicates the largest distortion.
On leaving the batch of crystals for 6 more days in CS2, a more dramatic structural rearrangement occurs, again in a SCSC manner, to form [Zn4O(L)3(DMF)]n (Figure (d)). One of the three crystallographically independent urea ligands undergoes rotation from the trans/trans conformation to the trans/cis arrangement, dramatically changing both the length and geometry of the linker (as well as its ability to bind guests or act as an organocatalyst) without altering the connectivity or interpenetration of the MOF (Citation 70 ). To accommodate the distortion, the SBU picks up a DMF molecule, leaving one of the Zn2+ centres in a 5-coordinate trigonal bipyramidal geometry (Figure (d)), very rarely seen in MOFs with the zinc acetate SBU, while the rest remain tetrahedral. This coordinated DMF molecule is likely one of those previously bound at the urea groups – only one of the three urea moieties now H-bonds a DMF guest – but exchange with the outer solvent cannot be excluded as a possibility. CS2 molecules fill the pores (Figure (a)).
Figure 6. (Colour online) (a) Portion of the crystal structure of [Zn4O(L)3(DMF)]n showing the presence of both CS2 and DMF in the pores. Disorder and H atoms removed for clarity. (b) The trans/trans conformation of L 2− , and (c) the trans/cis conformation of L 2− in the crystal structure of [Zn4O(L)3(DMF)]n. H atoms other than N-H units removed for clarity.
![Figure 6. (Colour online) (a) Portion of the crystal structure of [Zn4O(L)3(DMF)]n showing the presence of both CS2 and DMF in the pores. Disorder and H atoms removed for clarity. (b) The trans/trans conformation of L 2− , and (c) the trans/cis conformation of L 2− in the crystal structure of [Zn4O(L)3(DMF)]n. H atoms other than N-H units removed for clarity.](/cms/asset/de01bfb7-1c31-4131-bef3-bb25584ad640/gsch_a_1370095_f0006_oc.jpg)
The structure maintains a relatively cubic arrangement of the Zn4O SBUs (Figure (d)), despite the obvious deviation that occurs on rotation of the molecules of L 2− from the trans/trans conformation (Figure (b)) to the trans/cis conformation (Figure (c)). In fact, the angles around the SBUs show even less deviation from an ideal cube than the parent material (Table ), as the trans/cis linkers can still bridge two SBUs in an almost linear manner, despite their bent nature disposing the carboxylate units at an angle of approximately 120°, presumably generating the unusual mono-solvated Zn4O cluster.
The trans/cis linkers are, however, significantly shorter than the trans/trans linkers: the SBU-SBU distances measured between central μ4-O2– ligands is 17.55 Å for the trans/cis linker, as opposed to 19.68 Å and 19.94 Å for the two crystallographically independent trans/trans linkers, distances which are similar to those in the other Zn MOFs. The structure therefore is deviating from the cubic arrangement by pulling in two vertices on one side of the cube. We believe that the aforementioned flexibility of the material, derived from the L 2− ligands, allows it to endure such a significant change in linker length and geometry without breaking connectivity or altering interpenetration. This flexibility, combined with the labile coordination chemistry of the [Zn4O(RCO2)6] SBU, may, however, be responsible for the eventual framework collapse on drying – we were unable to successfully activate the porosity [Zn4O(L)3(DMF)2]n to ascertain if the urea units enhanced CO2 uptake.
Conclusions
We have exploited the flexibility of the urea-based linker, N,Nʹ-bis(4-carboxyphenyl)urea, to prepare three new framework materials with unusual SBUs: [Cd(L)(DMF)3]n, a one-dimensional coordination polymer; [Ca5(L)5(DMF)3(H2O)2]n a three-dimensional MOF linked by infinite calcium chains; and two solvates of [Zn4O(L)3(DMF)2]n, which has the prototypical IRMOF topology. In concordance with previous work, the urea moieties in [Cd(L)(DMF)3]n direct structure formation through interligand H-bonding, but the topological restrictions placed on the urea units in the other two materials leave them free to bind guest solvent molecules, and an independent report published during our study described [Zn4O(L)3(DMF)2]n to be an active organocatalyst as a result.
CS2 was used as a mimic to probe guest binding in [Zn4O(L)3(DMF)2]n, resulting in a number of single-crystal to single-crystal transformations that illustrated (i) the lability of the [Zn4O(RCO2)6] SBU, capturing crystallographic snapshots in unusual states of solvation, and (ii) the flexibility of the framework, enabled by the facile coordination chemistry of the cluster and the flexibility of the organic ligand. Three additional crystal structures showed the MOF breathing as it exchanged DMF pore solvent for CS2, eventually resulting in a dramatic configurational rearrangement of some of the urea units from the trans/trans to the trans/cis conformation, which are not capable of binding guests for organocatalysis. These results provide valuable structural insights into the dynamic behaviour of MOFs with flexible linkers upon activation, which have significant implications for the development of heterogeneous organocatalysts, and also give evidence for potential hydrolysis mechanisms. Additionally, we anticipate that CS2 may be considered a mild alternative solvent for activating MOFs in future; whilst [Zn4O(L)3(DMF)2]n did collapse on solvent removal, CS2 was the only solvent which penetrated its pores without damaging single crystals.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
The research was financially supported by Engineering and Physical Sciences Research Council (EPSRC) [grant number EP/L004461/1].
Acknowledgements
RSF thanks the Royal Society for receipt of a University Research Fellowship, and the University of Glasgow for funding. We thank the EPSRC UK National Crystallographic Service for single crystal data collection (Citation 40 ).
Related Research Data
References
- Furukawa, H. ; Cordova, K.E. ; O’Keeffe, M. ; Yaghi, O.M. Science 2013, 341 , 1230444.
- Kitagawa, S. ; Kitaura, R. ; Noro, S.-I. Angew. Chem. Int. Ed. 2004, 43 , 2334–2375.10.1002/(ISSN)1521-3773
- Férey, G. Chem. Soc. Rev. 2008, 37 , 191–214.10.1039/B618320B
- Inokuma, Y. ; Kawano, M. ; Fujita, M. Nature Chem. 2011, 3 , 349–358.10.1038/nchem.1031
- Otte, M. ACS Catal. 2016, 6 , 6491–6510.10.1021/acscatal.6b01776
- Alegre-Requena, J.V. ; Marqués-López, E. ; Herrera, R.P. ; Díaz, D.D. CrystEngComm 2016, 18 , 3985–3995.10.1039/C5CE02526E
- Rogge, S.M.J. ; Bavykina, A. ; Hajek, J. ; Garcia, H. ; Olivos-Suarez, A.I. ; Sepulveda-Escribano, A. ; Vimont, A. ; Clet, G. ; Bazin, P. ; Kapteijn, F. ; Daturi, M. ; Ramos-Fernandez, E.V. ; Llabres, I. ; Xamena, F.X. ; Van Speybroeck, V. ; Gascon, J. Chem. Soc. Rev. 2017, 46 , 3134–3184.
- Farrusseng, D. ; Aguado, S. ; Pinel, C. Angew. Chem. Int. Ed. 2009, 48 , 7502–7513.10.1002/anie.v48:41
- Gascon, J. ; Corma, A. ; Kapteijn, F. ; Llabrés, I. ; Xamena, F.X. ACS Catal. 2014, 4 , 361–378.10.1021/cs400959 k
- Liu, J. ; Chen, L. ; Cui, H. ; Zhang, J. ; Zhang, L. ; Su, C.-Y. Chem. Soc. Rev. 2014, 43 , 6011–6061.10.1039/C4CS00094C
- Ma, L. ; Wang, X. ; Deng, D. ; Luo, F. ; Ji, B. ; Zhang, J. J. Mater. Chem. A 2015, 3 , 20210–20217.10.1039/C5TA06248A
- Kitson, P.J. ; Marshall, R.J. ; Long, D. ; Forgan, R.S. ; Cronin, L. Angew. Chem. Int. Ed. 2014, 53 , 12723–12728.10.1002/anie.201402654
- Duan, J. ; Yang, Z. ; Bai, J. ; Zheng, B. ; Li, Y. ; Li, S. Chem. Commun. 2012, 48 , 3058–3060.10.1039/c2cc16231 h
- Park, J. ; Li, J.-R. ; Chen, Y.-P. ; Yu, J. ; Yakovenko, A.A. ; Wang, Z.U. ; Sun, L.-B. ; Balbuena, P.B. ; Zhou, H.-C. Chem. Commun. 2012, 48 , 9995–9997.10.1039/c2cc34622b
- Zheng, B. ; Yang, Z. ; Bai, J. ; Li, Y. ; Li, S. Chem. Commun. 2012, 48 , 7025–7027.10.1039/c2cc17593b
- Liu, B. ; Li, D.-S. ; Hou, L. ; Yang, G.-P. ; Wang, Y.-Y. ; Shi, Q.-Z. Dalton Trans. 2013, 42 , 9822–9825.10.1039/c3dt50997d
- Lee, C.-H. ; Huang, H.-Y. ; Liu, Y.-H. ; Luo, T.-T. ; Lee, G.-H. ; Peng, S.-M. ; Jiang, J.-C. ; Chao, I. ; Lu, K.-L. Inorg. Chem. 2013, 52 , 3962–3968.10.1021/ic302758 g
- Alsmail, N.H. ; Suyetin, M. ; Yan, Y. ; Cabot, R. ; Krap, C.P. ; Lü, J. ; Easun, T.L. ; Bichoutskaia, E. ; Lewis, W. ; Blake, A.J. ; Schröder, M. Chem. Eur. J. 2014, 20 , 7317–7324.10.1002/chem.201304005
- Paul, A. ; Karmakar, A. ; Guedes da Silva, M.F.C. ; Pombeiro, A.J.L. RSC Adv. 2015, 5 , 87400–87410.10.1039/C5RA14637B
- Karmakar, A. ; Desai, A.V. ; Manna, B. ; Joarder, B. ; Ghosh, S.K. Chem. Eur. J. 2015, 21 , 7071–7076.10.1002/chem.201406233
- Azhdari Tehrani, A. ; Esrafili, L. ; Abedi, S. ; Morsali, A. ; Carlucci, L. ; Proserpio, D.M. ; Wang, J. ; Junk, P.C. ; Liu, T. Inorg. Chem. 2017, 56 , 1446–1454.10.1021/acs.inorgchem.6b02518
- Roberts, J.M. ; Fini, B.M. ; Sarjeant, A.A. ; Farha, O.K. ; Hupp, J.T. ; Scheidt, K.A. J. Am. Chem. Soc. 2012, 134 , 3334–3337.10.1021/ja2108118
- Siu, P.W. ; Brown, Z.J. ; Farha, O.K. ; Hupp, J.T. ; Scheidt, K.A. Chem. Commun. 2013, 49 , 10920–10922.10.1039/c3cc47177b
- Dong, X.-W. ; Liu, T. ; Hu, Y.-Z. ; Liu, X.-Y. ; Che, C.-M. Chem. Commun. 2013, 49 , 7681–7683.10.1039/c3cc42531b
- Tehrani, A.A. ; Abedi, S. ; Morsali, A. ; Wang, J. ; Junk, P.C. J. Mater. Chem. A 2015, 3 , 20408–20415.10.1039/C5TA03835A
- Wang, X.-J. ; Li, J. ; Li, Q.-Y. ; Li, P.-Z. ; Lu, H. ; Lao, Q. ; Ni, R. ; Shi, Y. ; Zhao, Y. CrystEngComm 2015, 17 , 4632–4636.10.1039/C5CE00625B
- Ju, Z. ; Yan, S. ; Yuan, D. Chem. Mater. 2016, 28 , 2000–2010.
- Liu, W. ; Huang, X. ; Xu, C. ; Chen, C. ; Yang, L. ; Dou, W. ; Chen, W. ; Yang, H. ; Liu, W. Chem. Eur. J. 2016, 22 , 18769–18776.10.1002/chem.v22.52
- Rao, P.C. ; Mandal, S. ChemCatChem 2017, 9 , 1172–1176.10.1002/cctc.201601583
- Li, Q.-Y. ; Quan, Y. ; Wei, W. ; Li, J. ; Lu, H. ; Ni, R. ; Wang, X.-J. Polyhedron 2015, 99 , 1–6.10.1016/j.poly.2015.06.001
- Hall, E.A. ; Redfern, L.R. ; Wang, M.H. ; Scheidt, K.A. ACS Catal. 2016, 6 , 3248–3252.10.1021/acscatal.6b00424
- Dugan, E. ; Wang, Z. ; Okamura, M. ; Medina, A. ; Cohen, S.M. Chem. Commun. 2008, 3366–3368.10.1039/b806150e
- Luan, Y. ; Zheng, N. ; Qi, Y. ; Tang, J. ; Wang, G. Catal. Sci. Tech. 2014, 4 , 925–929.10.1039/c3cy00864a
- Zhang, X. ; Zhang, Z. ; Boissonnault, J. ; Cohen, S.M. Chem. Commun. 2016, 52 , 8585–8588.10.1039/C6CC03190K
- McGuirk, C.M. ; Katz, M.J. ; Stern, C.L. ; Sarjeant, A.A. ; Hupp, J.T. ; Farha, O.K. ; Mirkin, C.A. J. Am. Chem. Soc. 2015, 137 , 919–925.10.1021/ja511403t
- Cohen, S.M. ; Zhang, Z. ; Boissonnault, J.A. Inorg. Chem. 2016, 55 , 7281–7290.10.1021/acs.inorgchem.6b00828
- Forgan, R.S. ; Marshall, R.J. ; Struckmann, M. ; Bleine, A.B. ; Long, D.-L. ; Bernini, M.C. ; Fairen-Jimenez, D. CrystEngComm 2015, 17 , 299–306.10.1039/C4CE01379D
- Connon, S.J. Chem. Eur. J. 2006, 12 , 5418–5427.10.1002/(ISSN)1521-3765
- Zhang, Z. ; Schreiner, P.R. Chem. Soc. Rev. 2009, 38 , 1187–1198.10.1039/b801793j
- Coles, S.J. ; Gale, P.A. Chem. Sci. 2012, 3 , 683–689.10.1039/C2SC00955B
- Rigaku (2012). CrystalClear-SM Expert 3.1 b27, UK.
- Rigaku Oxford Diffraction (2015), CrysAlisPRO, UK.
- Bruker (2016). APEX3, Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2016). SAINT, Bruker AXS Inc., Madison, Wisconsin, USA.
- Sheldrick, G. Acta Cryst. A 2015, 71 , 3–8.10.1107/S2053273314026370
- Sheldrick, G. Acta. Cryst. C 2015, 71 , 3–8.10.1107/S2053229614024218
- Dolomanov, O.V. ; Bourhis, L.J. ; Gildea, R.J. ; Howard, J.A.K. ; Puschmann, H. J. Appl. Crystallogr. 2009, 42 , 339–341.10.1107/S0021889808042726
- Spek, A. Acta Cryst. C 2015, 71 , 9–18.10.1107/S2053229614024929
- Drewe, W.C. ; Nanjunda, R. ; Gunaratnam, M. ; Beltran, M. ; Parkinson, G.N. ; Reszka, A.P. ; Wilson, W.D. ; Neidle, S. J. Med. Chem. 2008, 51 , 7751–7767.10.1021/jm801245v
- Jayaramulu, K. ; Haldar, R. ; Maji, T.K. Polyhedron 2013, 52 , 553–559.10.1016/j.poly.2012.08.029
- Ruschewitz, U. ; Pantenburg, I. Acta Cryst. C 2002, 58 , 483–484.10.1107/S0108270102014658
- Chang, X.-H. ; Zhao, Y. ; Feng, X. ; Ma, L.-F. ; Wang, L.-Y. Polyhedron 2014, 83 , 159–166.10.1016/j.poly.2014.05.063
- Li, H. ; Eddaoudi, M. ; O’Keeffe, M. ; Yaghi, O.M. Nature 1999, 402 , 276–279.
- Han, S. ; Ma, Z. ; Wei, Y. ; Kravtsov, V.C. ; Luisi, B.S. ; Kulaots, I. ; Moulton, B. CrystEngComm 2011, 13 , 4838–4840.10.1039/c1ce05354j
- Kesanli, B. ; Cui, Y. ; Smith, M.R. ; Bittner, E.W. ; Bockrath, B.C. ; Lin, W. Angew. Chem. Int. Ed. 2005, 44 , 72–75.10.1002/(ISSN)1521-3773
- Yao, Q. ; Su, J. ; Cheung, O. ; Liu, Q. ; Hedin, N. ; Zou, X. J. Mater. Chem. 2012, 22 , 10345–10351.10.1039/c2jm15933c
- Das, M.C. ; Xu, H. ; Wang, Z. ; Srinivas, G. ; Zhou, W. ; Yue, Y.-F. ; Nesterov, V.N. ; Qian, G. ; Chen, B. Chem. Commun. 2011, 47 , 11715–11717.10.1039/c1cc12802 g
- Hobday, C.L. ; Marshall, R.J. ; Murphie, C.F. ; Sotelo, J. ; Richards, T. ; Allan, D.R. ; Düren, T. ; Coudert, F.-X. ; Forgan, R.S. ; Morrison, C.A. ; Moggach, S.A. ; Bennett, T.D. Angew. Chem. Int. Ed. 2016, 55 , 2401–2405.10.1002/anie.201509352
- Duan, J. ; Bai, J. ; Zheng, B. ; Li, Y. ; Ren, W. Chem. Commun. 2011, 47 , 2556–2558.10.1039/c0cc04146 g
- Wright, J.S. ; Metherell, A.J. ; Cullen, W.M. ; Piper, J.R. ; Dawson, R. ; Ward, M.D. Chem. Commun. 2017, 53 , 4398–4401.10.1039/C7CC01959A
- Wang, X. ; Jacobson, A.J. Micropor. Mesopor. Mater. 2016, 219 , 112–116.10.1016/j.micromeso.2015.07.038
- Nicole Power, K. ; Hennigar, T.L. ; Zaworotko, J.M. New J. Chem. 1998, 22 , 177–181.10.1039/a707640a
- Adams, C.J. ; Real, J.A. ; Waddington, R.E. CrystEngComm 2010, 12 , 3547–3553.10.1039/c0ce00149j
- Abrahams, B.F. ; Grannas, M.J. ; Hudson, T.A. ; Hughes, S.A. ; Pranoto, N.H. ; Robson, R. Dalton Trans. 2011, 40 , 12242–12247.10.1039/c1dt10962f
- Takamizawa, S. ; Nakata, E.-I. ; Saito, T. Chem. Lett. 2004, 33 , 538–539.10.1246/cl.2004.538
- Inokuma, Y. ; Matsumura, K. ; Yoshioka, S. ; Fujita, M. Chem. Asian J. 2017, 12 , 208–211.10.1002/asia.v12.2
- Ohba, M. ; Yoneda, K. ; Agustí, G. ; Muñoz, M.C. ; Gaspar, A.B. ; Real, J.A. ; Yamasaki, M. ; Ando, H. ; Nakao, Y. ; Sakaki, S. ; Kitagawa, S. Angew. Chem. Int. Ed. 2009, 48 , 4767–4771.10.1002/anie.v48:26
- Brozek, C.K. ; Michaelis, V.K. ; Ong, T.-C. ; Bellarosa, L. ; López, N. ; Griffin, R.G. ; Dincă, M. ACS Cent. Sci. 2015, 1 , 252–260.10.1021/acscentsci.5b00247
- Cychosz, K.A. ; Matzger, A.J. Langmuir 2010, 26 , 17198–17202.10.1021/la103234u
- Marshall, R.J. ; Griffin, S.L. ; Wilson, C. ; Forgan, R.S. J. Am. Chem. Soc. 2015, 137 , 9527–9530.10.1021/jacs.5b05434