
Abstract
Live bacterial therapeutics is gaining attention, especially for cancer therapy, because anaerobic bacteria selectively grow inside the solid tumours. However, the effect of tumour structure and bacterial characteristics on the pharmacokinetics of tumours is unclear; therefore, we aimed to elucidate the effects of tumour structure and types of bacteria on tumoral bacterial growth. Using six mouse xenograft models, including stroma-rich tumours similar to clinical tumours, and two models of live bacterial therapeutics, Salmonella typhimurium VNP20009 and Escherichia coli DH5α, we investigated bacterial growth and distribution in tumours after intravenous administration. Rapid growth of E. coli was observed in HCT116 and other tumours with few collagens, blood vessels not covered by mural cells, and a cancer cell area proliferated disorderly, whereas tumours with contrasting features, such as BxPC-3, showed lower bacterial growth and a limited intratumor distribution. Alternatively, Salmonella typhimurium VNP20009, when successfully proliferated (the probability was approximately 50%), grew to 108 colony forming units/g tissue even in BxPC-3 tumours, and its intratumor distribution was extensive. This study suggests that the development of new methods to modify tumour structure will be essential for the development of anti-tumour clinical therapies based on live bacterial therapeutics.
Introduction
The idea of using bacteria to treat cancers has been around for a long time, inspired by the fact that tumour growth is slower in some cases in cancer patients with bacterial infections. In modern medicine, research and development of bacteria-mediated cancer therapy began preclinical studies discovering intravenously administered anaerobic bacteria grow selectively in solid tumours. In fact, several studies have shown that various anaerobic bacteria administered intravenously in mice, including Clostridium [Citation1–3], Salmonella [Citation4], Bifidobacterium [Citation5, Citation6], and Escherichia [Citation7, Citation8], colonise and proliferate inside subcutaneously inoculated tumours. Interestingly, in various cases, bacterial administration induced significant tumour regression [Citation9, Citation10], suggesting that live bacterial therapeutics could be a future area for the research and development of cancer treatment. The anti-tumour effects of bacterial administration might be due to bacterial exotoxins and endotoxins that exert direct cytotoxicity against cancer cells and indirectly promote anticancer immunostimulation. However, these properties raise concerns about the adverse effects that might appear after systemic administration. Thus, attenuated strains for systemic inoculation, including Clostridium novyi-NT [Citation11], Salmonella typhimurium VNP20009 [Citation12], and Listeria vaccine [Citation13, Citation14], have been developed. Several clinical studies have been conducted using these strains, but unfortunately, in most cases these bacterial strains did not grow in tumours, and the anti-tumour effects were also very limited. The results of a phase I clinical trial of S. typhimurium VNP20009 inoculation in patients with metastatic melanoma or metastatic renal cell carcinoma showed no tumour regression, and only three of seven patients with doses greater than 3 × 108 CFU/m2 had viable bacteria in tumours, confirmed by biopsy [Citation15]. In another study, a 4-h infusion at a dose of 3 × 108 CFU/m2 did not produce any clinical effects, and the tumour biopsies contained S. typhimurium VNP20009 only in one of four patients on Day 5 after bacterial administration and none on Day 14 [Citation16]. The bacterial attenuation was recently thought to greatly weaken the bacterial antitumor effects as a trade-off; therefore, the current development of live bacterial therapeutics for cancer therapy is being focussed on designer bacteria, in which attenuated bacteria function as factories of anticancer substances to endow artificial, highly cancer-selective cytotoxicity and anticancer immunostimulation [Citation17, Citation18]. The discrepancies of bacterial growth in tumours between the pre-clinical and clinical studies may be due, in part, to the fact that the non-clinical studies used tumour models significantly different from the clinical tumours. Therefore, the bacterial growth and the anticancer effects in studies using animal models may not be properly evaluated.
The histological structure of tumours varies greatly depending on the type of cancer and its growth rate. The arrangement of tumour blood vessels, endothelial structure, and stroma abundance are especially relevant in anticancer drug delivery. In particular, refractory cancers such as pancreatic and gastric cancers are rich in the stroma, which acts as a physical barrier that complicates the delivery of many anticancer drugs to the core of the tumours. In preclinical studies on mice implanted with tumours subcutaneously and treated with several nanoparticular formulations, the number of nanoparticles and their distribution within tumours differed depending on the tumour type. For instance, polymeric micelles with a diameter of 100 nm were distributed throughout xenografted tumours of the colorectal cancer cell line CT26 with high vascularisation and slight stroma. In contrast, the polymeric micelles showed a limited distribution in xenografted tumours of the pancreatic cancer cell line BxPC-3 with abundant stroma. In addition, the physicochemical properties of the nanoparticles affected their distributions in tumours. In tumours of the pancreatic cancer cell line BxPC-3, particles with a diameter of 30 nm were more widely distributed than those with a diameter of 50–100 nm [Citation19]. These types of systematic investigations are essential for the clinical applications of live bacterial therapeutics; however, there have been virtually no such studies to date [Citation20–22].
This study aimed to elucidate the effects of tumour structure and characteristics of live bacterial therapeutics on their pharmacokinetics, especially in tumours. Due to the complex mechanisms of tumorigenesis, it is difficult to generate mouse xenograft models in which only one factor (for example, amount of tumoral stroma) is altered. Therefore, as realistically feasible for animal experiments, including from the perspective of animal welfare, and on a scale that would withstand systematic discussion, using six xenograft mouse models, including stroma-rich tumours that were similar to clinical tumours, we investigated bacterial survival and growth after intravenous administration as well as their distribution in tumours. The six tumour models used in this study were selected on the basis that they were as varied as possible, especially in terms of the amount of stroma, and that they have been used in the past in drug delivery and other related researches. In addition to S. typhimurium VNP20009, which has been clinically tested as described above [Citation15, Citation16, Citation23], we used Escherichia coli DH5α as a model for live bacterial therapeutics because it has been reported to be used for development of anticancer designer bacteria [Citation24, Citation25] and exhibit the standard survival and growth characteristics in xenografted tumours of common colorectal cancer and other cell lines [Citation7, Citation26, Citation27]. In addition, the changes in the tumour necrotic area and the levels of systemic inflammation after bacterial administration were evaluated.
Materials and methods
Cancer cell lines and animals
Human pancreatic cancer cell line BxPC-3, human lung cancer cell line Calu-3, human brain glioblastoma cell line U-87 MG, adenocarcinomic human alveolar basal epithelial cells A549, and human ovarian cancer cell line SK-OV-3 were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA), and the human colon cancer cell line HCT116 was obtained from RIKEN BioResource Research Centre (Tsukuba, Japan) through the National Bio-Resource Project of the MEXT, Japan. Calu-3 and U-87 MG cells were cultured in minimum essential medium, A549 cells were cultured in Ham’s F-12 medium with L-Gln, BxPC-3 cells were cultured in RPMI1640, HCT116 cells were cultured in Dulbecco’s Modified Eagle Medium, and SK-OV-3 cells were cultured in McCoy′s 5 A medium. Every medium was supplemented with 10% heat-inactivated foetal bovine serum and 1% (v/v) penicillin/streptomycin mix (Nacalai Tesque, Inc., Kyoto, Japan). All cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2.
Male athymic BALB/cCrSlc nude mice (6 weeks old) were purchased from Japan SLC, Inc. (Hamamatsu, Japan). Mice were subcutaneously inoculated into the right back flank with 3–8 × 106 cells in 50 μl of Hank’s balanced salt solution (Nacalai Tesque, Inc.). When the tumour volumes were approximately 200–800 mm3, the mice were ready for bacterial administration and other experiments. All animal experimental protocols were approved by the Ethics Committee on Animal Care and Use of the RIKEN Kobe Institute and were performed in accordance with the principles of laboratory animal care (NIH publication No. 85-23, revised 1985).
Bacterial culture and administration
E. coli DH5α competent cells, and ptdTomato and pZsGreen plasmids expressing the fluorescent proteins controlled by the pLac promoter, were purchased from Takara Bio Inc. (Kusatsu, Japan). Competent cells were transformed by the standard heat-shock method [Citation28]. E. coli cells were cultured in Lysogeny Broth (LB) medium containing 100 μg/ml carbenicillin with rotary shaking at 130 rpm at 37 °C. The cells were pre-cultured for 16–18 h till they reached the stationary phase, and then diluted 1/40, re-cultured for 2 h till an optical density of 0.7–0.9 at a wavelength of 600 nm (log phase) was obtained, and centrifuged at 6 000 × g at 25 °C for 3 min. The pellet was washed twice with and resuspended in saline. The solution was filtered using a MILLEX®-SV 5.0 μm syringe (Merck Millipore Ltd., Japan) immediately before administration.
An attenuated Salmonella typhimurium VNP20009 strain (ATCC 202165) was purchased from ATCC. The cells were cultured in modified LB medium (10 g/L Bacto Tryptone, 5 g/l Bacto Yeast Extract, 2 ml of 0.5 M CaCl2, and 2 ml of 0.5 M MgSO4/l, adjusted to pH 7 with 1 M NaOH) with rotary shaking at 130 rpm at 37 °C. The cells were pre-cultured overnight till they reached the stationary phase. They were diluted 1/20, re-cultured for 1 h till an optical density of 0.5–0.8 was obtained, and centrifuged (10 000 × g, 25 °C, 3 min). The pellet was washed twice with and resuspended in saline. The solution was diluted 1/10 immediately before administration with saline.
The colony forming units (CFUs) in the administered solution and tumour samples were determined as described previously [Citation27], except for the modified LB medium used for S. typhimurium VNP20009.
The DH5α/ptdTomato (2–7 × 107 CFU), DH5α/pZsGreen (1–5 × 107 CFU), and S. typhimurium VNP20009 (1–4 × 106 CFU) were injected into the tail vein of the xenograft mice under 1.5% isoflurane anaesthesia.
Haematoxylin and eosin (H&E) staining and aniline blue staining
Tumour tissues were dissected from euthanised mice and fixed with 4% paraformaldehyde (PFA) overnight. They were soaked in Dulbecco’s phosphate-buffered saline (DPBS; Nacalai Tesque, Inc.) and embedded in paraffin. Tissue samples were sectioned (3 μm thick cross sections) and stained with H&E following a standard protocol, except for the BxPC-3 tumour samples on Day 7 after S. typhimurium VNP20009 administration that were prepared as 7 μm-thick frozen sections as described below in Immunofluoerscence staining. Aniline blue staining was performed using the Trichrome Stain Kit (connective tissue stain) (Abcam, Cambridge, United Kingdom) without the haematoxylin staining step. All tumour specimens were scanned using a Leica SCN400 slide scanner (Leica Biosystems, Nussloch, Germany).
Analysis of collagen occupancy rate
Two sections (interval 200 μm) in the middle of each tumour (n = 3) were stained with aniline blue and scanned as described above. Collagen occupancy rate was analysed using Aperio ImageScope v12.4.0.7018 (Leica Biosystems) based on the following formula: collagen occupancy rate (%) = collagen area (blue)/whole slice (blue and red) × 100.
Immunofluorescence staining
Tumour samples were fixed with 4% PFA, embedded in an optimal cutting temperature compound (Tissue-Tek® O.C.T compound; SAKURA Finetek Japan Co., Ltd., Tokyo, Japan), and stored at −30 °C. Frozen sections 7 μm-thick were obtained with a cryostat (CryoStarTM NX70, Thermo Scientific) and immunostained with (i) Armenian hamster CD31 monoclonal primary antibody (1:100, MA3105, Thermo Fisher Scientific) and Alexa Fluor® 488-conjugated anti-Armenian hamster IgG secondary antibody (1:200, AB_2338997, Jackson ImmunoResearch), (ii) anti-alpha smooth muscle actin (ASMA) monoclonal antibody conjugated with Cy3 (1:100, C6198, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany), and anti-Salmonella antibody with FITC (1:200, ab69253, Abcam). Briefly, sections were washed with DPBS, incubated with blocking solution (DPBS with 1.5% BSA) at room temperature (RT, 20-22 °C) for 1 h, and then with primary antibodies diluted in the blocking solution at RT overnight. After 3 washes with DPBS, sections were incubated with the secondary antibody diluted in blocking solution at RT for 1 h or directly stained with 4′,6-diamidino-2-phenylindole (DAPI, 1:200 in blocking solution, #BS04, FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan). Finally, slides were coverslipped with ProLongTM Diamond Antifade Mountant (Thermo Fisher Scientific, Massachusetts USA).
Tumour tissue clearing with the ScaleCUBIC
The excised tumours from the euthanised mice were fixed with 4% PFA for no less than overnight and cut to 200–400 μm thickness using a vibratome (VT1200S, Leica Biosystems, Wetzlar, Germany). Next, sections were washed with DPBS, immersed twice in 50% ScaleCUBIC-1 (FUJIFILM Wako Pure Chemical Corporation) at RT for 30 min, and in 100% ScaleCUBIC-1 at 37 °C for 2-3 days (the solution was changed every day) until they were transparent. The samples were then washed twice in DPBS for 30 min, incubated with 50% ScaleCUBIC-2 (FUJIFILM Wako Pure Chemical Corporation) at RT for 30 min twice, and 100% ScaleCUBIC-2 at RT overnight. Finally, the cleared tissue samples were immersed in mounting solution (2:8; solution 1:solution 2, FUJIFILM Wako Pure Chemical Corporation), mounted on a 35 mm glass bottom dish (IWAKI, AGC Techno Glass Co., Ltd., Shizuoka, Japan) and coverslipped.
Fluorescence microscopy
The anti-CD31 or anti-ASMA immunolabeled sections were observed using laser scanning confocal microscopy (LSCM) using a Zeiss LSM710 microscope (Carl Zeiss Microscopy GmbH, Nussloch, Germany) with a 20× objective in a 7 μm depth setting. To image the whole cleared tissue slices from tumour samples of mice receiving E. coli, tile scans were acquired using the LSM710 microscope with a 10× objective in a 100 μm depth setting. The acquisition software used was ZEN 2 (blue edition) (Carl Zeiss Microscopy GmbH).
Images from immunolabeled sections of tumour samples from mice injected with S. typhimurium VNP20009 were acquired using an inverted LSCM microscope (Olympus) and an Andor Dragonfly 200 spinning disc using Fusion acquisition software. Whole section images and 9 tile high-magnification scan images were obtained with the 10× and 60× objectives, respectively. Each high magnification image was deconvolved, and the maximum intensity projection of the 6 μm image stack was obtained with the Imaris Viewer 9.7.0.
Analysis of ASMA occupancy rate
High-resolution images were analysed using the Zeiss ZEN 2 (blue edition) software. A total of 3–5 images were obtained from the anti-ASMA immunolabeled sections near the middle of each tumour, excluding the necrotic region, gland structure area, and the edge. Sections that were not immunolabeled were used to set the threshold value to remove the background signal. The ASMA occupancy rate was calculated based on the following formula: ASMA occupancy rate (%) = ASMA-positive pixel number/total pixel number × 100.
Enzyme-linked immunosorbent assay (ELISA) of mouse tumour necrosis factor-α (TNF-α)
Blood was collected from the inferior vena cava 1.5 h after bacterial administration and stored overnight at 4 °C. After centrifugation (15 000 × g, 4 °C, 10 min), the supernatant was collected as the serum. TNF-α concentrations in serum samples were determined using a Mouse TNF alpha Uncoated ELISA Kit (88-7324, Thermo Fisher Scientific) following the manufacturer’s instructions.
Statistical analyses
GraphPad Prism v. 7.03 (GraphPad Software, Inc., La Jolla, CA, USA) was used for statistical analyses. For parametric data comparison of three or more groups, one-way analysis of variance was performed, followed by Tukey’s multiple comparison test. For non-parametric data comparison of two groups, Mann-Whitney test was performed. For non-parametric data comparison of three or more groups, Kruskal-Wallis test was performed, followed by Dunn’s post hoc test. All P values were two-tailed, and differences were considered statistically significant for p < 0.05.
Results
Structure of subcutaneously xenografted tumours
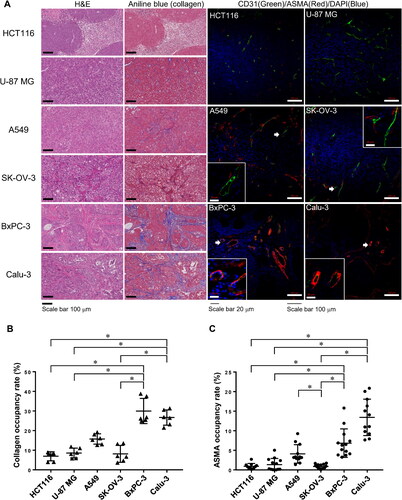
To evaluate the structure of subcutaneously xenografted tumours in mice, H&E and aniline blue staining as well as immunolabeling for the endothelial cell marker CD31 and fibroblast marker ASMA were performed in sections obtained from six tumour types (). In the H&E-stained sections, the HCT116 and U-87 MG tumours showed disordered growth of cancer cells with a central necrotic region. In contrast, the other four tumour types, especially BxPC-3 and Calu-3 tumours, showed a segregated islet-like distribution of cancer cells. In addition, in the aniline blue-stained sections, the BxPC-3 and Calu-3 tumours showed extensive aniline blue staining indicating the presence of collagen fibres, whereas HCT116, U-87 MG, A549, and SK-OV-3 tumours showed less staining (). The collagen occupancy rates of BxPC-3 and Calu-3 tumours were significantly higher than those of other tumours except for A549 tumours (). Moreover, immunostaining for CD31 and ASMA showed that the CD31-positive blood vessels in BxPC-3 and Calu-3 tumours presented a significantly higher ASMA occupancy rate than that in the HCT116, U-87 MG and SK-OV-3 tumours, which were almost not coated by ASMA-positive cells (). The A549 tumour showed an intermediate phenotype, with incomplete coverage by ASMA-positive cells.
Figure 1. Histological characteristics of subcutaneously xenografted tumours. (A) Representative images of H&E- and aniline blue staining in paraffin-embedded sections of HCT116 (colorectal carcinoma), U-87 MG (glioblastoma), A549 (lung carcinoma), SK-OV-3 (ovarian carcinoma), BxPC-3 (pancreatic carcinoma), and Calu-3 (non-small cell lung carcinoma) tumours. Scale bar, 100 μm. Representative confocal images and insets of CD31 (green) and ASMA (red) immunostaining of frozen sections of the different tumours, counterstained with DAPI (blue). Scale bars, 100 μm and 20 μm (insets). (B, C) Graphs showing the (B) collagen and (C) ASMA occupancy rates in different xenografted tumours. N = 3 mice per group (2-5 images per mouse); horizontal line and bars represent the mean ± SD for each group; Kruskal-Wallis test followed by Dunn’s post hoc test; *p < 0.05.
Differences in E. coli growth in xenografted tumours depending on the tumour types
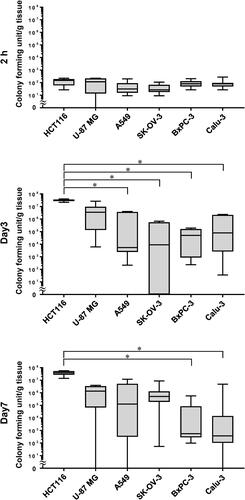
The number of E. coli (DH5α/ptdTomato) cells present in xenografted tumours after intravenous administration of the bacteria was analysed over time. The number of E. coli cells in the different tumours 2 h after administration accounted for less than 0.01% of the total number of bacterial cells administered, and there were no differences between tumour types (). However, the subsequent growth patterns of E. coli in the tumours differed depending on the tumour type. In the HCT116 and U-87 MG tumours, E. coli rapidly proliferated by Day 3, reaching median values of approximately 107 CFU/g and maintaining high levels thereafter. In the A549 and SK-OV-3 tumours, E. coli growth was slower, with median values of approximately 104 CFU/g and 105–106 CFU/g on Days 3 and 7, respectively. In contrast, in the BxPC-3 and Calu-3 tumours, the number of E. coli was 104–105 CFU/g on Day 3, and it decreased to 102 CFU/g on Day 7.
Figure 2. E. coli proliferation in subcutaneously xenografted tumours. Graphs show the viable bacteria concentration in HCT116, U-87 MG, A549, SK-OV-3, BxPC-3, and Calu-3 tumours of mice receiving 2.5–6.6 × 107 colony forming units of DH5α/ptdTomato intravenously. N = 5–9 mice per group; line within the box represents the median and whiskers represent the maximum and minimum values; Kruskal-Wallis test followed by Dunn’s post hoc test; *p < 0.05.
Distribution of E. coli in the tumours
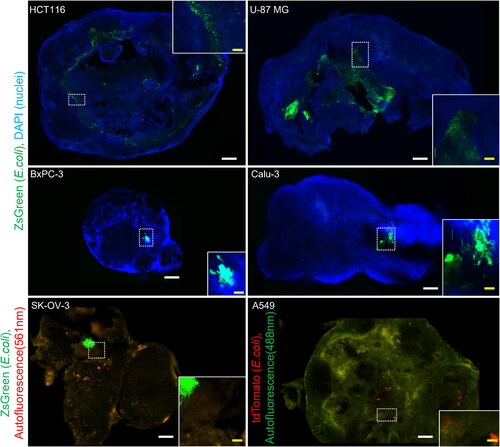
To analyse the distribution of bacteria in the tumours, confocal microscopy images were acquired from tumour samples collected 7 days after E. coli (DH5α/pZsGreen or DH5α/ptdTomato) administration (). In HCT116 tumours, the fluorescent signal from E. coli was mainly distributed within the necrotic region extending into the centre of the tumour and was more abundant closer to the viable region of the tumour edge. In U-87 MG tumours, E. coli was widely distributed in the centre of the tumour. In A549 and SK-OV-3 tumours, there were scattered clusters of E. coli and a sparse distribution of smaller populations. In contrast, no fluorescent signal from E. coli was observed in the majority of the BxPC-3 or Calu-3 tumour sections. In the few sections where the signal was observed, there were only one or two clusters; however, the sparse distribution of smaller E. coli populations seen in A549 and SK-OV-3 tumours was not observed.
Figure 3. Intratumoral E. coli distribution depending on subcutaneously xenografted tumours. Representative fluorescence images and insets of HCT116, U-87 MG, A549, SK-OV-3, BxPC-3, and Calu-3 cleared tumour samples from mice 7 days after intravenous injection of 1–5 × 107 colony forming units of DH5α/pZsGreen or 2–7 × 107 colony forming units of DH5α/ptdTomato. Sections of HCT116, U-87 MG, BxPC-3, and Calu-3 tumours were counterstained with DAPI. Scale bars, 1 mm and 200 μm (insets).
Growth and distribution of attenuated S. typhimurium VNP20009 in tumours
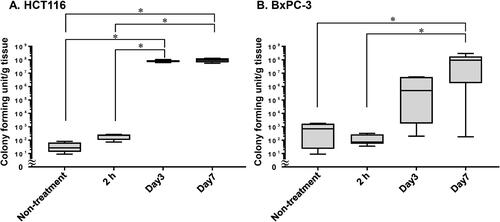
S. typhimurium VNP20009 was administered intravenously to mice previously implanted with HCT116 and BxPC3 tumours, which are typical of tumours where E. coli was more and less likely to grow, respectively, and the number of bacterial cells in the tumours was evaluated over time (). S. typhimurium VNP20009 cannot be selected with antibiotics, therefore, agar medium without antibiotics was used for CFU determination and some bacterial cells were detected in the control samples from HCT116 and BxPC-3 tumours of mice that did not receive S. typhimurium VNP20009. This experiment showed that the number of bacteria was between 101 and 103 CFU/g in the control group and 2 h after S. typhimurium VNP20009 administration. However, in HCT116 tumours, the number of bacteria increased rapidly on Day 3, reaching approximately 108 CFU/g, which was maintained until Day 7. In contrast, the number of bacteria increased gradually over time in BxPC-3 tumours, with a median value of 5 × 105 CFU/g on Day 3 and 108 CFU/g on Day 7. In addition, the data in and were reanalysed to investigate the difference in the numbers of bacteria in BxPC-3 tumours on Day 7 between S. typhimurium VNP20009 and E. coli. The difference was significant by Mann-Whitney test (p < 0.05, N = 11 and 8).
Figure 4. S. typhimurium VNP20009 proliferation in tumours. Time-dependent viable bacteria numbers in (A) HCT116 and (B) BxPC-3 tumours of mice receiving 1–4 × 106 colony forming units of S. typhimurium VNP20009 intravenously. N = 4–11 mice per group; line within the box represents the median and whiskers represent the maximum and minimum values; (A) 1-way ANOVA followed by Tukey post hoc test; (B) Kruskal-Wallis test followed by Dunn’s post hoc test; *p < 0.05.
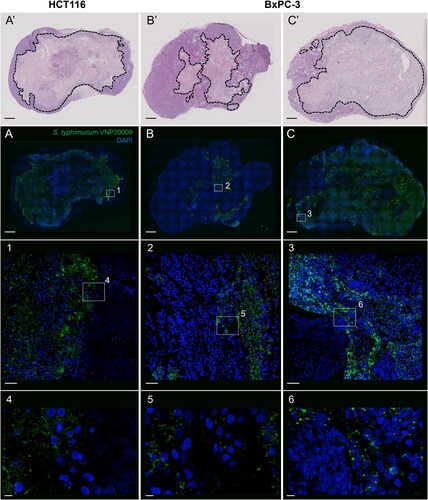
Confocal microscopy images of HCT116 tumour samples stained with FITC-labeled anti-Salmonella antibodies showed that S. typhimurium VNP20009 was massively distributed throughout the necrotic region in the centre of the tumours on Day 7 after administration (). In addition, small populations of S. typhimurium VNP20009 were observed frequently in the viable regions of the tumour margins. In BxPC-3 tumours, S. typhimurium VNP20009 formed numerous clusters in the necrotic area, and occasionally, distributions were observed in the glandular structures. However, only five of the nine BxPC-3 specimens showed a significant distribution of S. typhimurium VNP20009. The five tumours with larger necrotic areas contained more and larger bacterial colonies than those with smaller necrotic areas, which contained almost no bacteria.
Figure 5. Intratumoral S. typhimurium VNP20009 distribution in HCT116 and BxPC-3 tumours. (A-C) Representative maximum intensity projection of confocal fluorescence images and insets of immunostaining with anti-salmonella-FITC antibody (green) and counterstained with DAPI (blue) of HCT116 and BxPC-3 tumours of mice 7 days after the intravenous administration of 1–4 × 106 colony forming units of S. typhimurium VNP20009. (A’-C’) Images of H&E-stained adjacent sections of A-C. Scale bars, 1 mm (A-C and A’-C’), 50 μm (1-3), and 5 μm (4-6).
Expansion of tumour necrotic area after bacterial administration
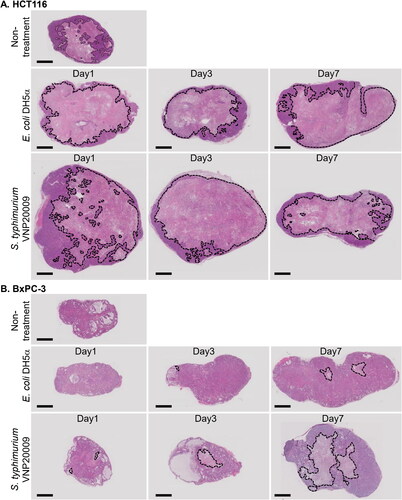
The changes in tumour necrotic area in HCT116 and BxPC-3 tumours after administration of DH5α/ptdTomato and S. typhimurium VNP20009 were evaluated by H&E staining (). These results showed that the necrotic area in HCT116 tumours was very similar after DH5α/ptdTomato and S. typhimurium VNP20009 administration. HCT116 tumours from mice not treated with bacteria presented vast necrotic areas in the centre and some viable areas in the periphery (). One day after administration of either bacterium, the necrotic areas extended to the edge of the tumours, and the viable areas became narrower. This morphology was maintained on Day 7. By contrast, the BxPC3 tumours had few, if any, necrotic regions and were quite small without bacterial administration (). After E. coli administration, small necrotic regions were observed on the first day, which gradually expanded over Days 3 and 7. Even so, the necrotic region was very limited on Day 7. Alternatively, after treatment with S. typhimurium VNP20009, the necrotic area was similar to that of E. coli on the first day, but on Days 3 and 7, there was a remarkable expansion of the necrotic area.
Figure 6. Expansion of the necrotic area in HCT116 and BxPC-3 tumours after bacterial administration. Images of H&E-stained equatorial sections of (A) HCT116 and (B) BxPC-3 tumours after intravenous administration of E. coli DH5α/ptdTomato (2–7 × 107 colony forming units, CFU) or S. typhimurium VNP20009 (1–4 × 106 CFU). Dotted lines indicate the necrotic regions. Scale bar, 2 mm.
Serum TNF-α concentrations after bacterial administration
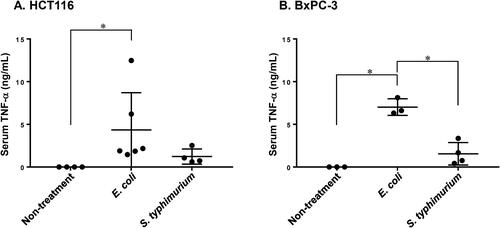
Increased permeability of tumour blood vessels caused by elevated TNF-α blood levels has been suggested to be one of the essential mechanisms of bacterial growth in CT26 tumours, which were rich in vessels and had little stroma [Citation29]. To investigate whether the same mechanism could explain the differences in growth between S. typhimurium VNP20009 and E. coli in BxPC-3 tumours, the TNF-α in serum concentration was measured 1.5 h after bacterial administration. In the absence of treatment, serum TNF-α levels were below the detection limit in mice implanted with HCT116 or BxPC-3 tumours (). In both groups of mice implanted with HCT116 or BxPC-3, there was a significant increase in TNF-α levels specifically after E. coli administration (). There was no effect of the type of tumour implanted on the increase in serum TNF-α.
Figure 7. Serum levels of TNF-α after administration of E. coli and S. typhimurium VNP20009. The serum TNF-α concentration in (A) HCT116 and (B) BxPC-3 tumour-xenografted mice was measured by ELISA 1.5 h after E. coli (2–7 × 107 colony forming units, CFU) or S. typhimurium VNP20009 (2 × 106 CFU) administration. N = 3–6 mice per group; horizontal line and bars represent mean ± SD. (A) Kruskal-Wallis test followed by Dunn’s post hoc test; (B) 1-way ANOVA followed by Tukey post hoc test; *P < 0.05.
Discussion
In this study, we evaluated the effects of systemic administration of bacteria in mice implanted with several types of xenografted tumours. Our results show that the colonisation, proliferation, and intratumoral localisation of anaerobic bacteria were significantly affected by the cancer type and bacterial species. In particular, in tumours with abundant collagen fibres and blood vessels tightly coated with mural cells, which are similar to clinical tumours, there was an extremely large variation in bacterial growth, which can be one of the challenges in using live bacterial therapeutics for cancer treatments.
Based on the results of the H&E and aniline blue staining and the CD31 and ASMA immunostainings, the six types of xenografted tumours used in this study were divided into the following three categories. In HCT116 and U-87 MG tumours, cancer cells proliferated in a disorderly manner, and there was no coating by the mural cells of the tumour blood vessels. In contrast, BxPC-3 and Calu-3 tumours were rich in collagen fibres, their blood vessels were well-covered by mural cells, and the cancer cell area was segmented into islets. A549 and SK-OV-3 tumours had intermediate characteristics, with few collagen fibres and incomplete coverage of blood vessels by mural cells, while the distribution of cancer cells was close to islet-like segmentation. These results are consistent with those of previous reports on subcutaneously xenografted tumours of each cell line [Citation30–36].
The rapid growth of E. coli DH5α in HCT116 and U-87 MG tumours by Day 3 and subsequent maintenance of their concentrations on Day 7 () are in agreement with many previous reports using mice subcutaneously bearing allogeneic tumours of mouse cancer cell lines, such as 4T1 and CT26 [Citation37–40], and immunodeficient mice with xenografted tumours, such as MCF-7, GI-101A, and F1A11 [Citation8,Citation39,Citation41]. After the proliferation under these conditions, bacterial concentrations in tumours were reported to be maintained over 2 weeks, although bacteria were almost completely eliminated from organs/tissues other than tumours by the hosts’ immune system in a few days. Semiquantitative evaluation of bacterial colonisation and growth in various tumours after intravenous administration has been reported using optical imaging, in which it was concluded that anaerobic bacteria colonised any tumour regardless of their origin [Citation41]. In this study, the other four tumour types (A549, SK-OV-3, BxPC-3, and Calu-3) showed low rates of survival and growth, and the variances were very high, especially in the BxPC-3 and Calu-3 tumours (). In a previous pharmacokinetic study using positron emission tomography, we showed that E. coli is cleared from the blood stream 5 min after intravenous administration [Citation27]. Therefore, the CFU in tumours measured 2 h after bacterial administration can be considered as the approximate number of E. coli reaching the tumours. These values were approximately 102 CFU/g tissue in the six tumours, suggesting that a very limited proportion of intravenously administered E. coli reached the tumours in all cases (0.3–0.5% injected dose/g, based on the previous PET study). Previously, it has been suggested that stable colony formation is critically determined by the presence of necrotic centres and stage of tumour development, rather than by age or tumour size [Citation41]. Interestingly, the results of the present study suggest that, more specifically, tumour structure determines whether E. coli can reach the tumours and proliferate.
The predominant distribution and scattering of E. coli around the central large necrotic area of the HCT116 tumours are in agreement with the distribution previously observed for E. coli K-12 and TOP10 and S. typhimurium SL7207 and VNP20009 in allogeneic subcutaneous CT26 and 4T1 tumours [Citation29, Citation37, Citation42, Citation43]. These tumours have similar structural characteristics to HCT116 tumours, with disordered growth of cancer cells and lack of coverage of tumour blood vessels by mural cells. It has been suggested that in CT26 tumours, neutrophils migrate in and surround the bacteria (S. typhimurium SL7207, E. coli TOP10), thus limiting the bacterial growth area within the tumours [Citation42]. The same mechanism is presumed to determine the distribution of bacteria within human cancer cell-derived xenografted tumours with similar structures to CT26 tumours. The widespread distribution of E. coli in the centre of U-87 MG tumours can be essentially due to the same mechanism as that in the HCT116 tumours, although the degree of neutrophil infiltration and the hypoxic environment suitable for E. coli growth may have been different. On the other hand, in the remaining four tumour types, the distribution of E. coli was very limited, suggesting the involvement of a completely different mechanism from that described above. Several hypotheses can be made about the cause of the limited distribution of E. coli in these tumours, which were rich in collagen fibres and presented blood vessels well-coated by mural cells and a cancer cell area segmented into islets. First, because of the mature and robust structure of these tumour blood vessels, it is possible that E. coli cannot migrate from the blood vessels to the tumour parenchyma and therefore can be easily eliminated by neutrophils and monocytes. Second, the islet-like segmentation of the cancer cell area might prevent E. coli from spreading. Third, unlike HCT116 and U-87 MG tumours, E. coli administration was unlikely to expand the necrotic area of BxPC-3 and Calu-3 tumours, which may be due to the lack of space for E. coli to grow. Recently, a phenomenon called dynamic vent has been reported, in which a nanoparticle ‘erupts’ through a temporary breach in a blood vessel [Citation44]. A similar phenomenon may occur in the intratumoral migration of E. coli. Future studies are required to investigate the exact mechanisms by which bacterial cells can reach and colonise in tumours.
S. typhimurium VNP20009, when successfully proliferated (however, the probability was approximately 50%), grew to 108 CFU/g tissue in BxPC-3 tumours by Day 7 after administration, and its intratumor distribution was extensive, unlike E. coli. These results suggest that the structural features of BxPC-3 tumours, such as abundant collagen fibres, vascular covering by mural cells, and islet-like segmentation of the cancer cell area, might also act as a structural/physical barrier to Salmonella growth by reducing the probability of extravasation and/or limiting the distribution area. However, once Salmonella reached the area where it could proliferate, it was more likely to grow than E. coli. This could be due to (i) Salmonella can form Salmonella-containing vacuole in host cells where it can survive and proliferate [Citation45, Citation46]; (ii) chemotaxis and motility are higher in Salmonella than in E. coli [Citation47–51]. These unique properties may allow Salmonella to overcome the barriers posed by tumours. However, there were not a few cases in which S. typhimurium VNP20009 did not grow in BxPC-3 tumours, and the distribution of S. typhimurium VNP20009 also varied with some cases being widespread and others being extremely limited.
It has been hypothesised that the increased permeability of tumour blood vessels induced by higher TNF-α blood level is one of the key mechanisms for bacterial growth in CT26 tumours, which are rich in blood vessels and nearly devoid of the stroma [Citation29]. In this study, the serum levels of TNF-α were similar or lower after administering S. typhimurium VNP20009 than those administering E. coli. Therefore, TNF-α was not the primary cause for the differences in their ability to grow on BxPC-3 tumours. However, there seems to be a correlation between bacterial growth and the extent of necrosis in BxPC-3 tumours after bacterial administration. This necrosis is probably caused by effects of S. typhimurium VNP20009 administration other than the influx of blood components into the tumour tissue. Elucidation of this induction mechanism will guide the design of future live bacterial therapeutics.
The results of this study suggest that the development of methods to modify the barriers in tumours is the key to the treatment of cancer through live bacterial therapeutics. These barriers are similar in many tumour-targeted drug delivery systems, such as nanoparticle formulations and antibody-drug conjugates, and methods to overcome them have been explored in some preclinical studies [Citation52–58]. Future studies are required to investigate whether any of these methods improves bacterial growth in BxPC-3 and other tumours in order to improve the clinical application of live bacterial therapeutics for cancer treatment.
To promote bacterial colonisation and growth in tumours rich in stroma, it is probably effective to loosen tumour vascular junction and decrease collagen and hyaluronan, and inhibit neutrophils and other cells from infiltrating into the tumours where bacteria have colonised. Specific methods can include pre-administration of collagenase, LY364947 (a transforming growth factor-β receptor inhibitor), or methylumbelliferone (an inhibitor of HA synthesis), and further attenuation of bacteria by shaving molecules involved in the induction of bacterial inflammation, such as lipopolysaccharide and flagellin.
This study strongly concludes that structures commonly found in clinical tumours, such as abundant collagen fibres and blood vessels tightly coated with mural cells, are among the factors that limit the growth of live bacterial therapeutics and their distribution area in tumours after intravenous administration, leading to high interindividual variability. Salmonella, which was evaluated in this study, is accounting for more than half of all research and development of anticancer live bacterial therapeutics probably due to the advantage of not forming spore [Citation14], and E. coli is also frequently being used. Therefore, this finding can be considered reasonably general. The following hypotheses are proposed as possible causes: (i) bacteria hardly migrate from blood vessels to the tumour parenchyma; (ii) bacteria are easily removed by neutrophils and monocytes; (iii) islet-like segmentation of the cancer cell area prevents the spread of bacteria; (iv) the necrosis after bacterial administration is rarely to occur and does not allow to create the space for bacterial growth. Currently, the development of designer bacteria for cancer therapy with enhanced cytotoxicity and immune-inducing activities, in addition to simple attenuated bacteria, is underway. This study suggests that the development of methods to modify the tumour structure is also required to effectively achieve anti-tumour effects with live bacterial therapeutics.
Authors’ contributions
Experimental design and investigation were performed by MT, KM, YW, and HM. Experiments and data analyses were carried by MT, EWS, SN, and AK. The manuscript was written, revised, and edited by MT, EWS, SN, AK, KM, YW, and HM. Supervision, Project administration, Funding acquisition was done by HM.
Acknowledgments
We would like to thank the RIKEN BioResource Research Center for providing HCT116 (RCB2979).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Parker RC, Plummer HC, Siebenmann CO, et al. Effect of histolyticus infection and toxin on transplantable mouse tumors. Proc Soc Exp Biol Med. 1947;66(2):461–467.
- Malmgren RA, Flanigan CC. Localization of the vegetative form of Clostridium tetani in mouse tumors following intravenous spore administration. Cancer Res. 1955;15(7):473–478.
- Lambin P, Theys J, Landuyt W, et al. Colonisation of clostridium in the body is restricted to hypoxic and necrotic areas of tumours. Anaerobe. 1998;4(4):183–188.
- Pawelek JM, Low KB, Bermudes D. Tumor-targeted Salmonella as a novel anticancer vector. Cancer Res. 1997;57(20):4537–4544.
- Yazawa K, Fujimori M, Amano J, et al. Bifidobacterium longum as a delivery system for therapy: selective localization and growth in hypoxic tumors. Cancer Gene Ther. 2000;7(2):269–274.
- Yazawa K, Fujimori M, Nakamura T, et al. Bifidobacterium longum as a delivery system for gene therapy of chemically induced rat mammary tumors. Breast Cancer Res Treat. 2001;66(2):165–170.
- Yu YA, Timiryasova T, Zhang Q, et al. Optical imaging: Bacteria, viruses, and mammalian cells encoding light-emitting proteins reveal the locations of primary tumors and metastases in animals. Anal Bioanal Chem. 2003;377(6):964–972.
- Yu YA, Shabahang S, Timiryasova TM, et al. Visualization of tumors and metastases in live animals with bacteria and vaccinia virus encoding light-emitting proteins. Nat Biotechnol. 2004;22(3):313–320.
- Mowday AM, Guise CP, Ackerley DF, et al. Advancing clostridia to clinical trial: past lessons and recent progress. Cancers (Basel). 2016;8(7):63.
- Zhou S, Gravekamp C, Bermudes D, et al. Tumour-targeting bacteria engineered to fight cancer. Nat Rev Cancer. 2018;18(12):727–743.
- Dang LH, Bettegowda C, Huso DL, et al. Combination bacteriolytic therapy for the treatment of experimental tumors. Proc Natl Acad Sci U S A. 2001;98(26):15155–15160.
- Clairmont C, Lee KC, Pike J, et al. Biodistribution and genetic stability of the novel antitumor agent VNP20009, a genetically modified strain of Salmonella typhimurium. J Infect Dis. 2000;181(6):1996–2002.
- Kim SH, Castro F, Gonzalez D, et al. Mage-b vaccine delivered by recombinant Listeria monocytogenes is highly effective against breast cancer metastases. Br J Cancer. 2008;99(5):741–749.
- Duong MTQ, Qin Y, You SH, et al. Bacteria-cancer interactions: bacteria-based cancer therapy. Exp Mol Med. 2019;51(12):1–15.
- Toso JF, Gill VJ, Hwu P, et al. Phase I study of the intravenous administration of attenuated Salmonella typhimurium to patients with metastatic melanoma. J Clin Oncol. 2002;20(1):142–152.
- Heimann DM, Rosenberg SA. Continuous intravenous administration of live genetically modified Salmonella typhimurium in patients with metastatic melanoma. J Immunother. 2003;26(2):179–180.
- Forbes NS. Engineering the perfect (bacterial) cancer therapy. Nat Rev Cancer. 2010;10(11):785–794.
- Riglar DT, Silver PA. Engineering bacteria for diagnostic and therapeutic applications. Nat Rev Microbiol. 2018;16(4):214–225.
- Cabral H, Matsumoto Y, Mizuno K, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6(12):815–823.
- Forbes NS, Coffin RS, Deng L, et al. White paper on microbial anti-cancer therapy and prevention. J Immunother Cancer. 2018;6(1):78.
- Pangilinan CR, Lee CH. Salmonella-Based targeted cancer therapy: updates on a promising and innovative tumor immunotherapeutic strategy. Biomedicines. 2019;7(2):36.
- Charbonneau MR, Isabella VM, Li N, et al. Developing a new class of engineered live bacterial therapeutics to treat human diseases. Nat Commun. 2020;11(1):1738.
- Nemunaitis J, Cunningham C, Senzer N, et al. Pilot trial of genetically modified, attenuated Salmonella expressing the E. coli cytosine deaminase gene in refractory cancer patients. Cancer Gene Ther. 2003;10(10):737–744.
- Afkhami-Poostchi A, Mashreghi M, Iranshahi M, et al. Use of a genetically engineered E. coli overexpressing β-glucuronidase accompanied by glycyrrhizic acid, a natural and anti-inflammatory agent, for directed treatment of Colon carcinoma in a mouse model. Int J Pharm. 2020;579:119159.
- Hosseini-Giv N, Bahrami AR, Matin MM. Application of bacterial directed enzyme prodrug therapy as a targeted chemotherapy approach in a mouse model of breast cancer. Int J Pharm. 2021;606:120931.
- Ryan RM, Green J, Lewis CE. Use of bacteria in anti-cancer therapies. Bioessays. 2006;28(1):84–94.
- Nomura S, Takahashi M, Kato AH, et al. Biosorption-based 64Cu-labeling of bacteria for pharmacokinetic positron-emission tomography. Int J Pharm. 2020;590:119950.
- Cohen SN, Chang AC, Hsu L. Nonchromosomal antibiotic resistance in bacteria: genetic transformation of Escherichia coli by R-factor DNA. Proc Natl Acad Sci U S A. 1972;69(8):2110–2114.
- Leschner S, Westphal K, Dietrich N, et al. Tumor invasion of Salmonella enterica serovar typhimurium is accompanied by strong hemorrhage promoted by TNF-alpha. PLoS One. 2009;4(8):e6692.
- Baker JHE, Kyle AH, Bartels KL, et al. Targeting the tumour vasculature: exploitation of low oxygenation and sensitivity to NOS inhibition by treatment with a hypoxic cytotoxin. PLoS One. 2013;8(10):e76832.
- Smith NR, Baker D, Farren M, et al. Tumor stromal architecture can define the intrinsic tumor response to VEGF-targeted therapy. Clin Cancer Res. 2013;19(24):6943–6956.
- Jiang Y, Allen D, Kersemans V, et al. Acute vascular response to cediranib treatment in human non-small-cell lung cancer xenografts with different tumour stromal architecture. Lung Cancer. 2015;90(2):191–198.
- Wegner CS, Hauge A, Simonsen TG, et al. DCE-MRI of sunitinib-induced changes in tumor microvasculature and hypoxia: a study of pancreatic ductal adenocarcinoma xenografts. Neoplasia. 2018;20(7):734–744.
- Park CR, Jo JH, Song MG, et al. Secreted protein acidic and rich in cysteine mediates active targeting of human serum albumin in U87MG xenograft mouse models. Theranostics. 2019;9(24):7447–7457.
- Gao Q, Yang Z, Xu S, et al. Heterotypic CAF-tumor spheroids promote early peritoneal metastatis of ovarian cancer. J Exp Med. 2019;216(3):688–703.
- Sato R, Imamura K, Semba T, et al. TGFβ signaling activated by cancer-associated fibroblasts determines the histological signature of lung adenocarcinoma. Cancer Res. 2021;81(18):4751–4765.
- Weibel S, Stritzker J, Eck M, et al. Colonization of experimental murine breast tumours by Escherichia coli K-12 significantly alters the tumour microenvironment. Cell Microbiol. 2008;10(6):1235–1248.
- Stern C, Kasnitz N, Kocijancic D, et al. Induction of CD4(+) and CD8(+) anti-tumor effector T cell responses by bacteria mediated tumor therapy. Int J Cancer. 2015;137(8):2019–2028.
- Kocijancic D, Felgner S, Schauer T, et al. Local application of bacteria improves safety of Salmonella-mediated tumor therapy and retains advantages of systemic infection. Oncotarget. 2017;8(30):49988–50001.
- Kang SR, Jo EJ, Nguyen VH, et al. Imaging of tumor colonization by Escherichia coli using 18F-FDS PET. Theranostics. 2020;10(11):4958–4966.
- Yu YA, Zhang Q, Szalay AA. Establishment and characterization of conditions required for tumor colonization by intravenously delivered bacteria. Biotechnol Bioeng. 2008;100(3):567–578.
- Westphal K, Leschner S, Jablonska J, et al. Containment of tumor-colonizing bacteria by host neutrophils. Cancer Res. 2008;68(8):2952–2960.
- Ganai S, Arenas RB, Sauer JP, et al. In tumors Salmonella migrate away from vasculature toward the transition zone and induce apoptosis. Cancer Gene Ther. 2011;18(7):457–466.
- Matsumoto Y, Nichols JW, Toh K, et al. Vascular bursts enhance permeability of tumour blood vessels and improve nanoparticle delivery. Nat Nanotechnol. 2016;11(6):533–538.
- Beuzón CR, Méresse S, Unsworth KE, et al. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. Embo J. 2000;19(13):3235–3249.
- Raman V, Van Dessel N, Hall CL, et al. Intracellular delivery of protein drugs with an autonomously lysing bacterial system reduces tumor growth and metastases. Nat Commun. 2021;12(1):6116.
- Kasinskas RW, Forbes NS. Salmonella typhimurium lacking ribose chemoreceptors localize in tumor quiescence and induce apoptosis. Cancer Res. 2007;67(7):3201–3209.
- Hansen CH, Endres RG, Wingreen NS. Chemotaxis in Escherichia coli: a molecular model for robust precise adaptation. PLoS Comput Biol. 2008;4(1):e1.
- Toley BJ, Forbes NS. Motility is critical for effective distribution and accumulation of bacteria in tumor tissue. Integr Biol (Camb). 2012;4(2):165–176.
- Silva-Valenzuela CA, Desai PT, Molina-Quiroz RC, et al. Solid tumors provide niche-specific conditions that lead to preferential growth of Salmonella. Oncotarget. 2016;7(23):35169–35180.
- Raman V, Van Dessel N, O’Connor OM, et al. The motility regulator flhDC drives intracellular accumulation and tumor colonization of Salmonella. J Immunother Cancer. 2019;7(1):44.
- Kano MR, Komuta Y, Iwata C, et al. Comparison of the effects of the kinase inhibitors imatinib, sorafenib, and transforming growth factor-β receptor inhibitor on extravasation of nanoparticles from neovasculature. Cancer Sci. 2009;100(1):173–180.
- Diop-Frimpong B, Chauhan VP, Krane S, et al. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci U S A. 2011;108(7):2909–2914.
- Zhang L, Nishihara H, Kano MR. Pericyte-coverage of human tumor vasculature and nanoparticle permeability. Biol Pharm Bull. 2012;35(5):761–766.
- Kohli AG, Kivimäe S, Tiffany MR, et al. Improving the distribution of doxil® in the tumormatrix by depletion of tumor hyaluronan. J Control Release. 2014;191:105–114.
- Miao L, Huang L. Exploring the tumor microenvironment with nanoparticles. Cancer Treat Res. 2015;166:193–226.
- Dolor A, Szoka FC. Digesting a path forward: the utility of collagenase tumor treatment for improved drug delivery. Mol Pharm. 2018;15(6):2069–2083.
- Xu S, Xu H, Wang W, et al. The role of collagen in cancer: from bench to bedside. J Transl Med. 2019;17(1):309.