
ABSTRACT
A Förster resonance energy transfer (FRET)-based assay was used to screen the FDA-approved compound library against the MERS-CoV helicase, an essential enzyme for virus replication within the host cell. Five compounds inhibited the helicase activity with submicromolar potencies (IC50, 0.73–1.65 µM) and ten compounds inhibited the enzyme with micromolar potencies (IC50, 19.6–502 µM). The molecular operating environment (MOE) was used to dock the identified inhibitors on the MERS-CoV helicase nucleotide binding. Strong inhibitors docked well in the nucleotide-binding site and established interactions with some of the essential residues. There was a reasonable correlation between the observed IC50 values and the MOE docking scores of the strong inhibitors (r2 = 0.74), indicating the ability of the in silico docking model to predict the binding of strong inhibitors. In silico docking could be a useful complementary tool used with the FRET-based assay to predict new MERS-CoV helicase inhibitors. The identified inhibitors could potentially be used in the clinical development of new antiviral treatment for MERS-CoV and other coronavirus related diseases, including coronavirus disease 2019 (COVID-19).
Introduction
Middle East Respiratory Syndrome (MERS), a highly pathogenic human coronavirus, was first identified in Saudi Arabia in 2012 [Citation1]. Since then, the disease spread to more countries in the Middle East and other regions of the world, including North Africa, Europe and North America [Citation2]. As of February 2019, 2279 laboratory-confirmed cases, including 806 deaths have been reported by the World Health Organization (WHO). The majority of these cases were reported in Saudi Arabia [Citation3]. Virus transmission from animals to humans was strongly linked to the direct interaction with dromedary camels (Camelus dromedarius) [Citation4]. Patients infected with MERS-CoV typically suffer from respiratory disease symptoms, including fever, cough, shortness of breath and to a lesser extent, gastrointestinal symptoms including diarrhoea and queasiness [Citation5]. Nearly 55% of the MERS-CoV patients require ICU care, and approximately 30% of the patients develop pleural effusions during the first few weeks of infection [Citation6].
Helicases are enzymes that use ATP to separate double-stranded nucleic acids (ds), producing two single-stranded nucleic acids (ss) [Citation7–9]. Helicases have been identified in the genomes of all three life kingdoms and in the genomes of many viruses [Citation10]. An estimated 31 DNA helicases and 64 RNA helicases are encoded in the human genome [Citation11]. The unwinding of the DNA through helicase activity is a central step in many cellular processes including replication, recombination, and DNA repair [Citation12]. Helicases are also essential for RNA transcription, mRNA splicing, mRNA export, translation, and RNA stability [Citation13–15]. Helicases are involved in molecular processes, including protein-nucleic acid complexes dissociation [Citation16–18], Holliday junction displacement [Citation19], chromatin remodelling [Citation20], and conformational modifications of nucleic acid [Citation21–23]. MERS-CoV is a positive single-stranded RNA (ssRNA) virus belonging to the Coronaviridae family, and has one of the largest known RNA genomes (~31.7 kb) [Citation24,Citation25]. The MERS-CoV genome is composed of ten open reading frames (ORFs). ORF1ab is located towards the 5′ end of the genome. After infection, a – 1 ribosomal frame shift during translation allows the synthesis of two large replicative polyproteins, pp1a (the ORF1a polyprotein) and pp1ab (the polyprotein made from ORF1a and ORF1b). The replicative polyproteins pp1a and pp1ab are proteolytically cleaved by the virus-encoded papain-like proteinase (PLpro) and 3 C-like protease (3CLpro) in 16 nonstructural proteins (nsps) [Citation26,Citation27]. The nsps are assembled into the membrane-associated replication-transcription complexes (RTCs). RTCs are responsible for viral genome replication and translation. RNA dependent RNA polymerase (nsp12) and helicase (nsp 13) are essential for RTCs functionality and ultimately for the whole life cycle of the virus [Citation28,Citation29]. As such, coronavirus helicases are seen as attractive therapeutic targets for inhibiting the replication of coronaviruses.
The Severe Acute Respiratory Syndrome CoV (SARS-CoV) helicase (S-nsp13), COVID-19 helicase and MERS-CoV helicase (M-nsp13) can unwind dsDNA and dsRNA in a 5′-to-3′ direction [Citation23,Citation30,Citation31]. The kinetic parameters of recombinant M-nsp13 have been determined and are similar to the parameters of S-nsp13 [Citation23]. The crystal structure of the MERS-CoV helicase reveals that it is a multiple-domain protein similar to nsp 13 of all other nidoviruses. It has an N-terminal Cys/His rich domain (CH) associated with three zinc atoms, a C-terminal SF1 helicase core that contains two RecA-like subdomains, and a beta-barrel domain [Citation32]. The MERS-CoV nsp13 crystal structure indicates that residues Gln404, Arg443 and Arg567 form hydrogen bonds with a sulphate moiety that is possibly involved in NTP hydrolysis. The residue Tyr442 group contributes to the stabilization of the adenosine base [Citation32]. Several S-nsp13 and M-nsp13 inhibitors have been identified using Förster resonance energy transfer (FRET)-based microplate screening assays [Citation25,Citation33,Citation34]. Nsp13 inhibitors can either inhibit enzyme unwinding activity or inhibit its ATPase activity [Citation35]. In this study, we are reporting several new M-nsp13 inhibitors identified through a FRET-based microplate screening assay of the commercially available FDA-approved list.
Several studies emphasize the advantage of using the MOE in silico docking as a tool to predict several helicase inhibitors, including the COVID-19 helicase, Zika virus helicase and hepatitis C virus helicase [Citation36–38]. Despite its computational advantages, the in silico docking ability to predict helicase inhibitors remains limited and the need for direct measurement of these inhibitors continues to be essential [Citation36]. In the current study, the in silico docking model was used as complementary to the FRET-based assay to identify new MERS-CoV helicase inhibitors.
Materials and methods
The Tocriscreen library of marketed FDA-approved compounds (Tocris Biosciences, Bristol, United Kingdom), the natural product screening library (Selleck Chemicals, Houston, USA), and microfluor 2 black U-bottom 96-well plates (Fisher Scientific) were used for the FRET screening. The DNA substrate fork was made of black hole quencher (BQH) modified oligonucleotides: 5′-TCACCACCACGTATCTGAGCCTGGGCGA(BHQ)-3′ and fluorescein modified oligonucleotide: 5′-(FLUORESCEIN)TCGCCCAGGCTCAGATACGACCACCACT-3′. The RNA substrate fork was made of 5′-UCACCACCACGUAUCUGAGCCUGGGCGA(BHQ)-3′ and 5′-(FLUORESCEIN)UCGCCCAGGCUCAGAUACGACCACCACU-3′oligonucleotides. All oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA).
Expression and purification
The MERS-CoV nsp13 (M-nsp13) bacterial expression plasmid was donated by Dr. Adeyemi O. Adedeji while he was at Midwestern University, Glendale, Arizona, USA. The M-nsp13 cDNA was cloned into the pET-52b vector with a Strep-tag at its N-terminus (Strep-tag-pET52b-M-nsp13) [Citation23]. M-nsp3 was expressed and purified as described previously [Citation33]. Briefly: pET-52b-M-nsp13 plasmids were transformed into BL21 (DE3) cells, which were grown overnight at 37°C in a starter non-inducing media (50 mM Na2HPO4, 50 mM NH4Cl, 5 mM Na2SO4, 2 mM MgSO4.7H2O, 10 μM FeCl3.6H3O, 4 μM CaCl2, 2 μM Mn3.4H3O, 2 μM ZnSO4.7H3O, 0.4 μM CoCl2.6H3O, 0.4 μM CuCl2.2H3O, 0.4 μM NiCl2.6H3O, 0.4 μM Na2MoO4.5H3O, Na2SeO3.5H3O, 0.4 μM H3BO3, 0.5% glucose, 0.25% aspartate, and 100 μg/ml ampicillin). Cells are transferred at a 1:1000 dilution into expression ZYM-5052 auto inducing media (10 g/L N-Z amine, 5 g/L yeast extract, 50 mM Na2HPO4, 50 mM NH4Cl, 5 mM Na2SO4, 2 mM MgSO4.7H2O, 10 μM FeCl3.6H3O, 4 μM CaCl2, 2 μM Mn3.4H3O, 2 μM ZnSO4.7H3O, 0.4 μM CoCl2.6H3O, 0.4 μM CuCl2.2H3O, 0.4 μM NiCl2.6H3O, 0.4 μM Na2MoO4.5H3O, Na2SeO3.5H3O, 0.4 μM H3BO3, 0.5% glycerol, 0.05% glucose, 0.2% alpha-lactose, and 100 μg/ml ampicillin) and left to grow at 37°C while shaking at 210 rpm for 5 h until the solution was turbid. The temperature was lowered to 20°C and shaken overnight until the OD600 plateau. The cells were harvested through centrifugation at 4000 × g for 10 min and washed once with lysis buffer:100 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5% glycerol, 0.1% Triton X, 5 mM beta-mercaptoethanol (or 1 M DTT), 0.15 mg/ml lysozyme, 1 mM phenylmethylsulfonyl fluoride (PMSF). The pellet was resuspended in 100 ml lysis buffer. The cell suspension was sonicated on ice at 75% amplitude for 15:15 s pulse for 2 min. The lysate was centrifuged at 17,500 × g for 45 min. The supernatant fluid was applied to a Strep-Tactin column previously equilibrated with two volumes of washing buffer: 100 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5% glycerol, 0.1% Triton X, and 5 mM beta-mercaptoethanol (or 1 M DTT). After the cell lysate completely passed through the column, the column was washed 5 times with 1 column volume of washing buffer and eluted with the same buffer containing 10 mM desthiobiotin. The eluate dialysed overnight in the washing buffer. M-nsp13 containing fractions were concentrated and stored at −80°C.
Helicase/ATPase activity assay
Throughout the purification procedure, enzyme activity was detected by measuring ATPase activity in samples using the E. coli DNA Helicase ATPase Assay Kit (ProFoldin, Hudson, MA). Briefly: 20 nM M-nsp13 is incubated with 250 nM dsDNA and 0.25 mM ATP in a reaction buffer containing 20 mM HEPES, pH 7.5, 20 mM potassium glutamate, 1 mM DTT, 0.005% Triton X-100, 10 mM MgCl2 at room temperature for 60 min in total volume, 30 µl. Released inorganic phosphate was detected by adding 45 µl detection dye to the reaction mixture in a 1:1.5 ratio. After 5 min incubation, absorbance of the solution was measured at 650 nm. The protein concentration was measured with Bradford’s assay reagent and the purity of the sample determined with SDS-PAGE. The positive control (E. coli helicase) is provided with the kit.
FRET-based screening assay in 96-well plates
The helicase dsDNA and dsRNA substrates were prepared by annealing a fluorescein-labelled oligonucleotide with a black hole quencher (BHQ)-labelled oligonucleotide at 1:1.2 ratio in 50 mM Tris, NaCl 50 mM, pH 8.0 in total volume 150 µl [Citation33]. The mixture was first heated to 95°C for 5 min and then cooled slowly to room temperature. The commercially available FDA-approved library of compounds (Tocriscreen Library of Marketed FDA-Approved Compounds) were lyophilized and added to 96-well plates. The source plates were prepared by dissolving the compounds in 100% v/v dimethyl sulphoxide (DMSO) to a final concentration of 10 mM. Each compound was assayed at a 100 µM final concentration by adding 1 µl of each compound from the source plate (10 mM) to the final assay volume of 100 µl. For the subsequent assays, the sub-plates of several factors dilutions (in 100% DMSO v/v) were prepared from the source plates. All the plates were stored at –80°C. The fluorescence due to helicase activity was measured in black 96-well flat-bottom corning plates. The assay volume for each well was 100 µl containing final concentrations of 20 mM HEPES, 20 mM NaCl, 5 mM MgCl2, 0.01% bovine serum albumin (BSA), 2 mM DTT, 5% glycerol, 50 nM nsp13, 0.5 mM ATP, 75 nM fluorescein- and black hole quencher-labelled dsDNA, 2 µM trap, unlabelled single-stranded DNA complementary to the BHQ-labelled DNA strand, and different final concentrations of inhibitors. A volume of 50 µl enzyme solution of the assay buffer containing 100 nM nsp13, 1 mM ATP, and 4 µM trap was preincubated with 1 µl of different concentrations of the inhibitors at room temperature for 5 min, and the reactions were initiated by adding 50 µl of assay buffer containing 150 nM labelled dsDNA or dsRNA. The reactions were allowed to proceed for 10 min at 30°C. The samples were excited at wavelength 495 nm and fluorescence emission was measured at wavelength 520 nm using a plate-reader (SpectraMax M5, Molecular Devices, USA).
Enzyme kinetics and IC50 determinations
The dsDNA and dsRNA substrates were prepared by annealing a fluorescein-labelled oligonucleotide with a black hole quencher as previously mentioned. To determine the Km values for the DNA and RNA, the helicase activity was measured by the FRET-based assay, as previously mentioned, with various concentrations of dsDNA or dsRNA (0 nM, 5 nM, 10 nM, 20 nM, 40 nM, 80 nM and 160 nM). To determine the Km values for the ATP, the helicase activity was measured with the FRET-based assay as previously mentioned, using dsDNA or dsRNA as the first substrate with various concentrations of ATP (0 mM, 0.25 mM, 0.5 mM, 1.5 mM, 3 mM, and 6 mM). Data were fitted to the Michaelis-Menten curve using GraphPad Prism. IC50 determinations for the inhibition of helicase activity were performed using varying concentrations of each inhibitor (0 µM, 1 × 10−3 µM, 1.7 × 10−3 µM, 5.1 × 10−3 µM, 15.2 × 10−3 µM, 45.7 × 10−3 µM, 0.1372 µM, 0.4115 µM, 1.234 µM, 3.704 µM, 11.11 µM, 33.33 µM, and 100 µM). The data were fitted to a 4-parameter logistic fit for the dose-response curves using GraphPad Prism.
Active site modelling
The in silico study was performed using a published crystal structure (PDB_ID: 5WWP) [Citation32]. The MERS-CoV helicase x-ray structure (3.0 Å) revealed a SO42- anion in the nucleotide-binding pocket. Amino acids Gln404, Arg443, Arg567 and Tyr442 were found to be essential for sulphate moiety stabilization [Citation32]. MOE (MOE, 2019.0102; Chemical Computing Group ULC, Canada) was used to dock the 15 molecules identified by the FRET screening into the nucleotide-binding pocket of the MERS-CoV helicase, allowing the active site to be flexible around 4.5 Å from the ligand. Each molecule was allowed seven final poses which interacted with the essential amino acids. Only the poses that contributed to the interaction with the key amino acids were included in the final set of molecules. The correlation between the docking scores and the observed IC50 was calculated.
Results
MERS-CoV helicase purification and activity
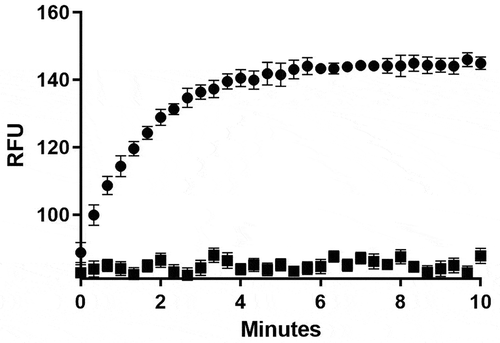
After expression in BL21 (DE3) cells, the MERS-CoV helicase was purified using the Strep-Tactin column, about 1 mg of nsp13 was produced from 1 L media. To validate the purified MERS-CoV helicase activity, both ATPase and helicase activities were detected. The ATPase activity was determined by measuring the total amount of released inorganic phosphate (). The helicase activity was detected by measuring the emission at 520 nm after excitation at 495 nm (). Assay conditions and a standard curve of the single-stranded RNA and DNA labelled with fluorescein were established to convert RFUs to change in double strand concentration – see Figures S1, S2 (supplementary material).
Figure 1. MERS-CoV helicase ATPase activity measured by detecting the change in absorbance of the released inorganic phosphate in the presence of no enzyme (□), 20 nM positive control E. coli helicase (![]()
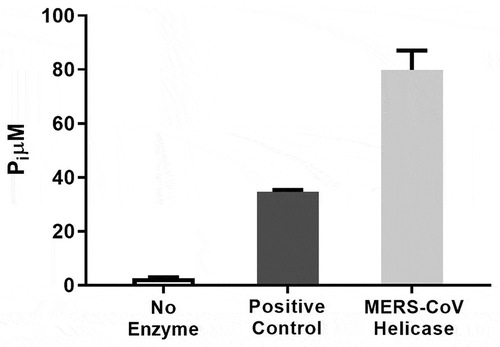
Figure 2. MERS-CoV helicase unwinding activity measured by detecting the relative change in fluorescence units (RFU) of the separated fluorescein-labelled ssRNA (excitation 495 nm, emission 520 nm) in the presence of no enzyme (■) or 50 nM MERS-CoV helicase (●)
MERS-CoV kinetic properties
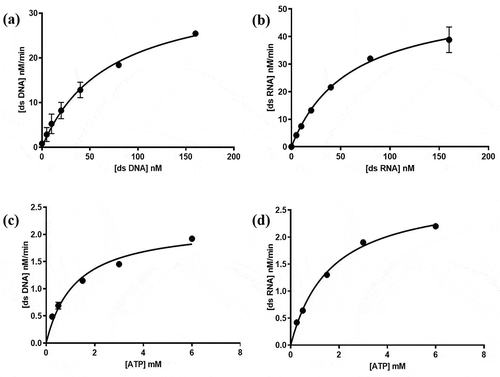
To compare the MERS-CoV helicase unwinding of dsDNA and dsRNA substrates, the kinetic parameters for the MERS-CoV helicase unwinding activity were measured using the FRET-based assay. Michaelis-Menten constants and maximum velocities were measured for dsDNA and dsRNA substrates, and ATP was used as a second substrate – see ). The same parameters were measured for the ATP, and the dsDNA or dsRNA was used as a second substrate – see ). The kinetic parameters values are displayed in .
Table 1. Kinetic parameters of MERS-CoV helicase
Figure 3. Michaelis-Menten curves of MERS-CoV helicase unwinding activity established by FRET-based assay using different concentrations of (a) dsDNA substrate, (b) dsRNA substrate, (c) ATP when dsDNA used as second substrate, and (d) ATP when dsRNA used as second substrate. Error bars represent standard deviation of triplicate samples
First round screening for MERS-CoV helicase activity inhibitors
The first round screening was performed in 100 µM final concentration conditions. In total, 1014 compounds from the Marketed FDA-Approved library and 124 compounds of the Natural Product library were screened using a FRET-based assay in 96-well plates to determine their ability to inhibit MERS-CoV helicase activity. Fifteen compounds of the FDA-Approved library inhibited the MERS-CoV helicase activity at 100 µM. No inhibitors were detected from the Natural Product library.
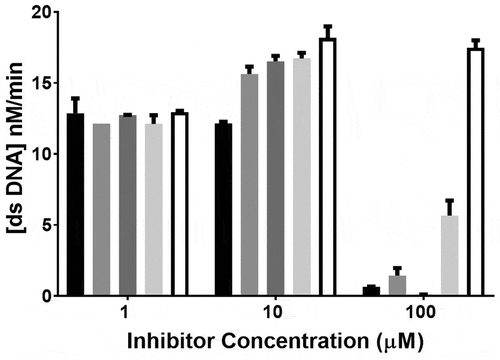
A second round of screening was performed to determine the inhibitory effect of the 15 compounds at 1 µM, 10 µM and 100 µM final concentrations. The assay for each compound concentration (1, 10 and 100 µM) was performed in a separate plate and each included DMSO as control. The MERS-COV helicase activity was clearly observed in the DMSO well serving as the control with some variation between the plates. The four compounds (doxorubicin HCl, epirubicin HCl, mitoxantrone 2HCl and daunorubicin HCl) yielded almost complete inhibition of the MERS-CoV helicase at 10 µM and 100 µM concentrations – see . Idarubicin largely inhibited the MERS-CoV helicase at 10 µM and 100 µM concentrations with relatively less inhibition at 100 µM. Ten compounds (otilonium bromide, caspofungin acetate, tolcapone, sunitinib malate, ethacridine lactate monohydrate, bazedoxifene HCl, masitinib, ruxolitinib, raloxifene HCl, and diminazene aceturate) yielded almost complete inhibition of MERS-CoV at 10 µM. Selected results are shown – see .
Figure 4. MERS-CoV helicase unwinding activity measured by FRET-based assay using different concentrations of strong inhibitors: idarubicin HCl (■), doxorubicin HCl (![]()
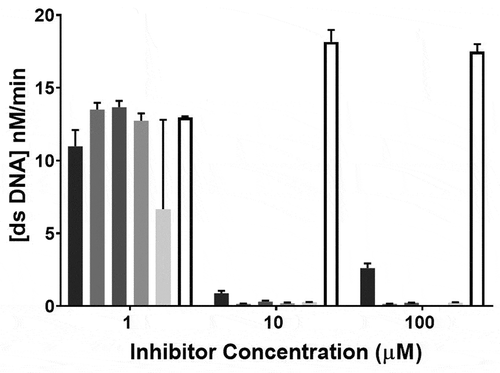
Figure 5. MERS-CoV helicase unwinding activity was measured by FRET-based assay using different concentrations of weak inhibitors: sunitinib malate (■), bazedoxifene HCl (![]()
IC50 determinations
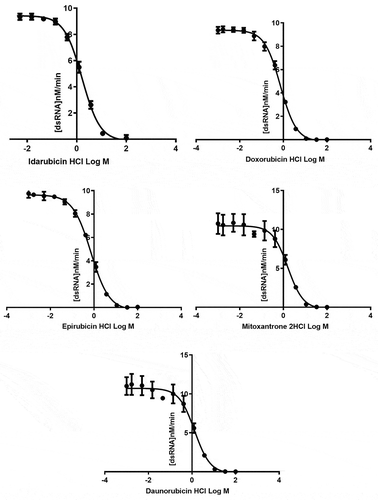
The MERS-CoV helicase can unwind both dsDNA and dsRNA. During the infection process, the MERS-CoV uses the dsRNA as a template. The kinetic parameters for the MERS-CoV helicase unwinding of the dsDNA and dsRNA were measured, and compared with the FRET-based assay. The MERS-CoV helicase can use dsDNA and dsRNA equivalently – see . The dsRNA was used to measure the IC50 of the inhibitors to mimic a natural substrate. To validate the inhibitory effect of the inhibitors, the IC50 value of each compound was determined using the FRET-based assay. The IC50 values of idarubicin HCl, doxorubicin HCl, epirubicin HCl, mitoxantrone 2HCl and daunorubicin HCl were in the lower micromolar range (≤2 μM) – see . In contrast, the IC50 values for otilonium bromide, caspofungin acetate, tolcapone, sunitinib malate, ethacridine lactate monohydrate, bazedoxifene HCl, masitinib, ruxolitinib, raloxifene HCl, and diminazene aceturate were in the high micromolar range (20–500 μM) – see .
Table 2. IC50 of MERS-CoV inhibitors
Figure 6. Dose-response curves of MERS-CoV helicase activity established by FRET-based assay using different concentrations of idarubicin HCl, doxorubicin HCl, epirubicin HCl, mitoxantrone 2HCl and daunorubicin HCl. Error bars represent standard deviation of triplicate samples
Active site modelling
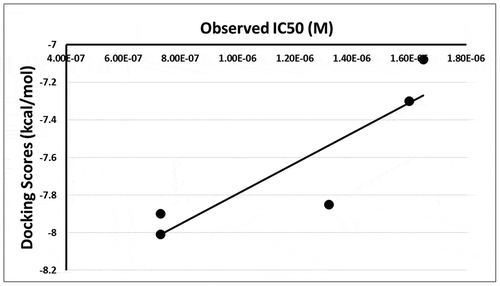
The 15 inhibitor compounds detected by the FRET-assay were conformationally docked into the nucleotide-binding pocket using the MOE, in which seven final poses were allowed for each molecule while the protein was fixed. Fifty molecules, including the FRET detected strong inhibitors, had posed with multiple interactions with several amino acids. The docking scores, interactions and the predicted IC50 values of the strong and weak inhibitors were calculated from the observed IC50 values – see . The Doxorubicin HCl, the best docking molecule, interactions with the MERS-CoV helicase are displayed in . Docking maps of all the inhibitors are displayed in Figures S3 – S11 (supplementary material). The correlation (r2 = 0.78) found between the observed IC50 values and the docking scores of the strong inhibitors are displayed in . Although the weak inhibitors, sunitinib malate, bazedoxifine HCl, masitinib, raloxifene HCl and ruxolitinib produced good docking scores: −7.32, −7.89, −9.12 and −6.55 respectively, they did not have many interactions within the active site. Sunitinib malate and ruxolitinib showed no residue interactions within the active site. The weak inhibitors otilonium bromide, caspofungin acetate, tolcapone, ethacridine lactate monohydrate, and diminazene aceturate did not fit in the binding site, and no docking scores were reported. Finally, no correlation was found between the observed IC50 values and the docking scores of the weak inhibitors (r2 = 0.34) or between all the inhibitors’ observed IC50 values and docking scores (r2 = 0.01) – see Figures S12 and S13 (supplementary material).
Table 3. Predicted IC50, docking scores and interactions of MERS-CoV of all inhibitors
Figure 7. (a) Molecular docking of doxorubicin HCl to MERS-CoV helicase (PDB_ID: 5WWP) binding site allowing protein 4.5 Å flexibility away from ligand, (b) multiple interactions of doxorubicin HCl with essential amino acid: Arg443 and Arg567, interaction map
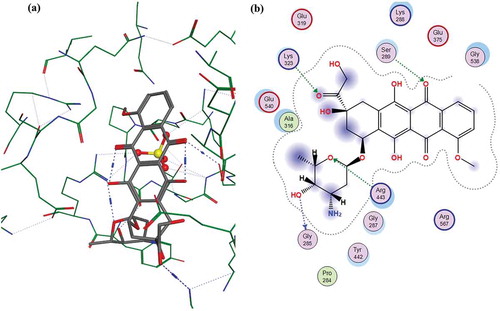
Figure 8. Correlation of observed IC50’s and docking scores of strong inhibitors
Discussion
The MERS-CoV helicase is essential in the formation and function of RTCs, responsible for the replication of MERS-CoV RNA and subsequently, the formation of new viruses in the host cell [Citation39]. This central role in the process of MERS-CoV infection makes it a possible antiviral target. In previous studies, a FRET-based screening assay was used to identify SARS-CoV helicase inhibitors [Citation33]. This study is the first study to report using recombinant MERS-COV helicase in a FRET-based screening assay to identify new potential MERS-CoV inhibitors by screening FDA-approved and the natural product libraries. The merit of MERS-CoV inhibitors detected within the marketed FDA-approved library is that they can rapidly progress to clinical trials.
Previous studies indicated that the SARS-CoV helicase takes a longer time to unwind longer double-strand nucleic acids. The SARS-helicase unwinds nucleic acid at a rate of ~280 bp s-1 with a preference to binding to a fork conformation. The MERS-CoV showed a similar preference [Citation33]. The length and design of the substrate (dsRNA or dsDNA) used in the assay are designed to allow complete separation under the effect of helicase activity. The helicase assay conditions used have no effect of FRET-based measurements [Citation33].
The size and activities of the M-nsp13 expressed in the BL21 (DE3) bacterial system were similar to those reported in literature [Citation23,Citation32]. M-nsp13 has two activities: helicase activity and ATPase activity. Similar to literature, the expressed M-nsp13 unwind both dsDNA and dsRNA [Citation23,Citation33]. The kinetic parameters Km and Vmax indicated that the MERS-CoV helicase could use dsDNA and dsRNA equally, as shown by the Km values of both substrates. Similar behaviour was reported for both MERS-CoV [Citation23] and SARS-CoV [Citation30] helicases. The use of dsDNA or dsRNA as a substrate with different ATP concentrations did not affect the rate of ATP hydrolysis, as shown by the similar Km values. The obtained Km values for the ATP are in agreement with literature [Citation23,Citation32]. Since the MERS-CoV helicase unwinds dsRNA during the natural infection process [Citation14], dsRNA substrate was used for the IC50 value determination.
The two rounds of screening in this study revealed two categories of inhibitors: strong inhibitors that inhibit at sub-micromolar concentrations and relatively weak inhibitors that require higher concentrations to inhibit helicase activity. The observed DMSO variation when the compounds were tested at different concentrations (1, 10 and 100 uM) could be attributed to the fact that the assay for each concentration was performed in a separate plate in triplicate. Small molecules can possibly inhibit helicase by competing with either the ATP or double-strand nucleic acid [Citation86]. These molecules could non-competitively inhibit helicase by binding to sites other than the active site. However, it should be noted that chelating the nucleic acid by a small molecule would also show an apparent inhibition behaviour, similar to the competitive inhibition effect. The binding constants of doxorubicin, epirubicin, daunorubicin, mitoxantrone and idarubicin to DNA were reported (3.4 × 104 M−1, 1.27 × 106 M−1, 2.04 × 106 M−1, 5.0 × 106 M−1, 5.14 × 105 M−1 respectively) [Citation55,Citation87–89]. The strong binding between these molecules and DNA could explain their strong apparent inhibition of the helicase. The values of the IC50 determinations of the strong inhibitors, except doxorubicin, are close or within the range of a reported dissociation constant. Variations could be attributed to different assay conditions, substrate composition and enzyme preparation. The close values of the IC50 values and the reported dissociation constants support the assumption that these compounds are affecting the MERS-CoV helicase by chelating the dsRNA substrate. Despite previous argument, the possibility of the binding of these inhibitors to the helicase in a competitive or non-competitive manner cannot be completely excluded. When docked to the active site, all the strong inhibitors docked with reasonably good docking scores. Since these strong inhibitors are all anthracycline-based structures, the close values of the docking score are expected. Some of these inhibitors, particularly doxorubicin, daunorubicin and idarubicin showed possible interactions with the residue Arg443 and a sulphate moiety, essential for enzyme activity [Citation32]. It is true that the current assay may be limited in discriminating the mode of inhibition of the strong inhibitors, whether they are competing with the substrate over the active site or directly chelating the substrate itself, however, this assay remains valid for the weak inhibitors that directly bind to the enzyme. The weak inhibitor mechanism, inhibition of the nsp13 helicase by binding to the enzyme itself, is supported by the fact that none of these weak inhibitors has been reported to bind double-strand nucleic acids. When docked to the enzyme active site, the weak inhibitors either did not fit well in the active site (otilonium bromide, caspofungin acetate, tolcapone, ethacridine lactate monohydrate, and diminazene aceturate) or docked well but with no clear interactions with any of the essential residue were detected, except masitinib which has one detected interaction with the essential residue Arg567 – see Figure S8 (supplementary material).
The observed correlation between the IC50 values and the docking scores of the strong inhibitors detected by the FRET-assay suggests that in silico docking of molecules is possibly an efficient tool to screen different molecule libraries. The calculated IC50 values of the strong inhibitors from the docking scores, using the correlation equation, were very close to the directly measured IC50 values. However, this model is limited when it comes to the weak inhibitors since there is no clear correlation between the docking scores and the weak inhibitors’ IC50 values. This poor correlation can be explained by the wide diversity of the molecular structures of these molecules. This model can discriminate weak inhibitors that do not fit in the active site; however, it can falsely predict weak inhibitors as strong inhibitors if they happen to fit in the active site. This shortcoming of the model emphasizes the need to use this docking tool as a complementary tool to direct FRET assay. The docking model can be used to exclude weak inhibitors from possible strong inhibitors, and the strong inhibitors can be confirmed by the direct FRET assays.
Due to the unavailability of a BCL3 laboratory, the inhibitors have not been tested to inhibit the replication of live MERS-CoV in cells. However, they represent good candidates for testing, not only for MERS-CoV live virus, but also for SARS-COV and other Coronaviruses, and are good starting molecules for further optimization. The inhibitors with lower potencies, such as otilonium bromide, caspofungin acetate, tolcapone, sunitinib malate, masitinib, bazedoxifene HCl, and ethacridine lactate monohydrate, could be optimized to develop as strong MERS-CoV helicase inhibitors. Finally, future x-ray crystallography studies could shed more light on how these molecules interact with the helicase and provide some guidance for the development of stronger inhibitors.
Supplemental Material
Download MS Word (4.7 MB)Acknowledgements
We thank KAIMRC for funding this research work. We thank all KAIMRC researchers and administrative staff in assisting directly and indirectly in this research. We thank the University of Midwestern for donating the MERS-CoV helicase expression plasmid (nsp13).
Disclosure statement
The authors declare that they have no conflict of interests.
Availability of data and materials
All the data used to support the findings of this study are included in the article and supplementary information is provided in the supplementary section. In addition, the data used to support the findings of this study are available from the corresponding author on request.
Supplementary material
Supplementary data for this article can be accessed at: https://doi.org/10.1080/1062936X.2020.1857437
Additional information
Funding
References
- A.M. Zaki, S. Van Boheemen, T.M. Bestebroer, A.D.M.E. Osterhaus, and R.A.M. Fouchier, Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia, N. Engl. J. Med. 367 (2012), pp. 1814–1820. doi:10.1056/NEJMoa1211721.
- A. Chafekar and B.C. Fielding, MERS-CoV: Understanding the latest human coronavirus threat, Viruses 10 (2018), pp. 93. doi:10.3390/v10020093.
- A. Mubarak, W. Alturaiki, and M.G. Hemida, Middle East respiratory syndrome coronavirus (MERS-CoV): Infection, immunological response, and vaccine development, J. Immunol. Res. (2019), pp. 6491738. doi:10.1155/2019/6491738.
- E.I. Azhar, S.A. El-Kafrawy, S.A. Farraj, A.M. Hassan, M.S. Al-Saeed, A.M. Hashem, and T.A. Madani, Evidence for camel-to-human transmission of MERS coronavirus, N. Engl. J. Med. 370 (2014), pp. 2499–2505. doi:10.1056/NEJMoa1401505.
- V.S. Raj, A.D.M.E. Osterhaus, R.A.M. Fouchier, and B.L. Haagmans, MERS: Emergence of a novel human coronavirus, Curr. Opin. Virol. 5 (2014), pp. 58–62. doi:10.1016/j.coviro.2014.01.010.
- K.M. Das, E.Y. Lee, S.E. Al Jawder, M.A. Enani, R. Singh, L. Skakni, N. Al-Nakshabandi, K. AlDossari, and S.G. Larsson, Acute Middle East respiratory syndrome coronavirus: Temporal lung changes observed on the chest radiographs of 55 patients, Am. J. Roentgenol. 205 (2015), pp. W267–W274. doi:10.2214/AJR.15.14445.
- T.M. Lohman, Escherichia coli DNA helicases: Mechanisms of DNA unwinding, Mol. Microbiol. 6 (1992), pp. 5–14. doi:10.1111/j.1365-2958.1992.tb00831.x.
- T.M. Lohman and K.P. Bjornson, Mechanisms of helicase-catalyzed DNA unwinding, Annu. Rev. Biochem. 65 (1996), pp. 169–214. doi:10.1146/annurev.bi.65.070196.001125.
- T.M. Lohman, Helicase-catalyzed DNA unwinding, J. Biol. Chem. 268 (1993), pp. 2269–2272.
- G.W. Owttrim, RNA helicases: Diverse roles in prokaryotic response to abiotic stress, RNA Biol. 10 (2013), pp. 96–110. doi:10.4161/rna.22638.
- R.M. Brosh and S.W. Matson, History of DNA helicases, Genes (Basel) 11 (2020), pp. 255. doi:10.3390/genes11030255.
- K. Geider and H. Hoffmann-Berling, Proteins controlling the helical structure of DNA, Annu. Rev. Biochem. 50 (1981), pp. 233–260. doi:10.1146/annurev.bi.50.070181.001313.
- S. Lain, J.L. Riechmann, and J.A. Garciá, RNA helicase: A novel activity associated with a protein encoded by a positive strand RNA virus, Nucleic Acids Res. 18 (1990), pp. 7003–7006. doi:10.1093/nar/18.23.7003.
- Z. Xing, W.K. Ma, and E.J. Tran, The DDX5/Dbp2 subfamily of DEAD-box RNA helicases, Wiley Interdiscip. Rev. RNA 10 (2019), pp. e1519. doi:10.1002/wrna.1519. 2.
- V. Khemici and P. Linder, RNA helicases in RNA decay, Biochem. Soc. Trans. 46 (2018), pp. 163–172. doi:10.1042/BST20170052.
- A.K. Byrd and K.D. Raney, Displacement of a DNA binding protein by Dda helicase, Nucleic Acids Res. 34 (2006), pp. 3020–3029. doi:10.1093/nar/gkl369.
- C.S. Jordan and S.W. Morrical, Regulation of the bacteriophage T4 Dda helicase by Gp32 single-stranded DNA-binding protein, DNA Repair (Amst.) 25 (2015), pp. 41–53. doi:10.1016/j.dnarep.2014.10.002
- S. Aarattuthodiyil, A.K. Byrd, and K.D. Raney, Simultaneous binding to the tracking strand, displaced strand and the duplex of a DNA fork enhances unwinding by Dda helicase, Nucleic Acids Res. 42 (2014), pp. 11707–11720. doi:10.1093/nar/gku845.
- J. Xia, L.T. Chen, Q. Mei, C.H. Ma, J.A. Halliday, H.Y. Lin, D. Magnan, J.P. Pribis, D.M. Fitzgerald, H.M. Hamilton, M. Richters, R.B. Nehring, X. Shen, L. Li, D. Bates, P.J. Hastings, C. Herman, M. Jayaram, and S.M. Rosenberg, Holliday junction trap shows how cells use recombination and a junction-guardian role of RecQ helicase, Sci. Adv. 2 (2016), pp. e1601605. doi:10.1126/sciadv.1601605.
- B. Bartholomew, ISWI chromatin remodeling: One primary actor or a coordinated effort, Curr. Opin. Struct. Biol. 24 (2014), pp. 150–155. doi:10.1016/j.sbi.2014.01.010.
- R. Lestini and B. Michel, UvrD controls the access of recombination proteins to blocked replication forks, Embo J. 26 (2007), pp. 3804–3814. doi:10.1038/sj.emboj.7601804.
- O. Cordin, J. Banroques, N.K. Tanner, and P. Linder, The DEAD-box protein family of RNA helicases, Gene 367 (2006), pp. 17–37. doi:10.1016/j.gene.2005.10.019.
- A.O. Adedeji and H. Lazarus, Biochemical characterization of Middle East respiratory syndrome coronavirus helicase, mSphere 1 (2016), pp. e00235–16. doi:10.1128/mSphere.00235-16.
- N. Zhang, S. Jiang, and L. Du, Current advancements and potential strategies in the development of MERS-CoV vaccines, Expert. Rev. Vaccines 6 (2014), pp. 761–774. doi:10.1586/14760584.2014.912134.
- M. Alnazawi, A. Altaher, and M. Kandeel, Comparative genomic analysis MERS CoV isolated from humans and camels with special reference to virus encoded helicase, Biol. Pharm. Bull. 40 (2017), pp. 1289–1298. doi:10.1248/bpb.b17-00241
- V. Thiel, K.A. Iva, Á. Putics, T. Hertzig, B. Schelle, S. Bayer, B. Weißbrich, E.J. Snijder, H. Rabenau, H.W. Doerr, A.E. Gorbalenya, and J. Ziebuhr, Mechanisms and enzymes involved in SARS coronavirus genome expression, J. Gen. Virol. 84 (2003), pp. 2305–2315. doi:10.1099/vir.0.19424-0.
- L. Subissi, I. Imbert, F. Ferron, A. Collet, B. Coutard, E. Roly, and B. Canard, SARS-CoV ORF1b-encoded nonstructural proteins 12-16: Replicative enzymes as antiviral targets, Antiviral Res. 101 (2014), pp. 122–130. doi:10.1016/j.antiviral.2013.11.006.
- L. Subissi, C.C. Posthuma, A. Collet, J.C. Zevenhoven-Dobbe, A.E. Gorbalenya, E. Roly, E.J. Snijder, B. Canard, and I. Imbert, One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities, Proc. Natl. Acad. Sci. U S A 111 (2014), pp. E3900–E3909. doi:10.1073/pnas.1323705111.
- E. Prentice, J. McAuliffe, X. Lu, K. Subbarao, and M.R. Denison, Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins, J. Virol. 78 (2004), pp. 9977–9986. doi:10.1128/JVI.78.18.9977-9986.2004.
- K.A. Iva, V. Thiel, J.C. Dobbe, Y. van der Meer, E.J. Snijder, and J. Ziebuhr, Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase, J. Virol. 78 (2004), pp. 5619–5632. doi:10.1128/JVI.78.11.5619-5632.2004.
- J.A. Tanner, R.M. Watt, Y.B. Chai, L.Y. Lu, M.C. Lin, J.S. Peiris, L.L. Poon, H.F. Kung, and J.D. Huang, The severe acute respiratory syndrome (SARS) coronavirus NTPas/helicase belongs to a distinct class of 5′ to 3′ viral helicases, J. Biol. Chem. 278 (2003), pp. 39578–39582. doi:10.1074/jbc.C300328200.
- W. Hao, J.A. Wojdyla, R. Zhao, R. Han, R. Das, I. Zlatev, M. Manoharan, M. Wang, and S. Cui, Crystal structure of Middle East respiratory syndrome coronavirus helicase, PLoS Pathog. 13 (2017), pp. e1006474. doi:10.1371/journal.ppat.1006474.
- A.O. Adedeji, K. Singh, N.E. Calcaterra, M.L. DeDiego, L. Enjuanes, S. Weiss, and S.G. Sarafianos, Severe acute respiratory syndrome coronavirus replication inhibitor that interferes with the nucleic acid unwinding of the viral helicase, Antimicrob. Agents Chemother. 56 (2012), pp. 4718–4728. doi:10.1128/AAC.00957-12.
- A.O. Adedeji, K. Singh, A. Kassim, C.M. Coleman, R. Elliott, S.R. Weiss, M.B. Frieman, and S.G. Sarafianos, Evaluation of SSYA10-001 as a replication inhibitor of severe acute respiratory syndrome, mouse hepatitis, and Middle East respiratory syndrome coronaviruses, Antimicrob. Agents Chemother. 58 (2014), pp. 4894–4898. doi:10.1128/AAC.02994-14.
- F. Ganzina, 4′-epi-doxorubicin, a new analogue of doxorubicin: A preliminary overview of preclinical and clinical data, Cancer. Treat. Rev. 10 (1983), pp. 1–22. doi:10.1016/S0305-7372(83)80029-2.
- J.F. Borgio, H.S. Alsuwat, W.M. Al Otaibi, A.M. Ibrahim, N.B. Almandil, L.I. Al Asoom, M. Salahuddin, B. Kaaj, and S. AbdulAzeez, State-of-the-art tools unveil potent drug targets amongst clinically approved drugs to inhibit helicase in SARS-CoV-2, Arch. Med. Sci. 16 (2020), pp. 508–518. doi:10.5114/aoms.2020.94567.
- S.L. Badshah, N. Ahmad, A. Ur Rehman, K. Khan, A. Ullah, A. Alsayari, A.B. Muhsinah, and Y. Mabkhot, Molecular docking and simulation of Zika virus NS3 helicase, BMC Chem. 13 (2019), pp. 67. doi:10.1186/s13065-019-0582-y.
- A. Wadood, M. Riaz, R. Uddin, and Z. Ul-Haq, In silico identification and evaluation of leads for the simultaneous inhibition of protease and helicase activities of HCV NS3/4A protease using complex based pharmacophore mapping and virtual screening, PLoS One 9 (2014), pp. e89109. doi:10.1371/journal.pone.0089109.
- P. Durai, M. Batool, M. Shah, and S. Choi, Middle East respiratory syndrome coronavirus: Transmission, virology and therapeutic targeting to aid in outbreak control, Exp. Mol. Med. 47 (2015), pp. e181. doi:10.1038/emm.2015.76.
- L.D. Williams, C.A. Frederick, G. Ughetto, and A. Rich, Ternary interactions of spermine with DNA: 4′-epiadriamycin and other DNA: Anthracycline complexes, Nucleic Acids Res. 18 (1990), pp. 5533–5541. doi:10.1093/nar/18.18.5533.
- E.R. Podell, D.J. Harrington, D.J. Taatjes, and T.H. Koch, Crystal structure of epidoxorubicin-formaldehyde virtual crosslink of DNA and evidence for its formation in human breast-cancer cells, Acta Crystallogr. D. Biol. Crystallogr. 55 (1999), pp. 1516–1523. doi:10.1107/s0907444999008161.
- J.P. Overington, B. Al-Lazikani, and A.L. Hopkins, How many drug targets are there, Nat. Rev. Drug Discov. 5 (2006), pp. 993–996. doi:10.1038/nrd2199.
- P. Imming, C. Sinning, and A. Meyer, Drugs, their targets and the nature and number of drug targets, Nat. Rev. Drug Discov. 5 (2006), pp. 821–834. Erratum in: Nat. Rev. Drug Discov. 6 (2007) pp. 126. doi:10.1038/nrd2132.
- T. Petit, M. Wilt, M. Velten, R. Millon, J.F. Rodier, C. Borel, R. Mors, P. Haegelé, M. Eber, and J.P. Ghnassia, Comparative value of tumour grade, hormonal receptors, Ki-67, HER-2 and topoisomerase II alpha status as predictive markers in breast cancer patients treated with neoadjuvant anthracycline-based chemotherapy, Eur. J. Cancer 40 (2004), pp. 205–211. doi:10.1016/s0959-8049(03)00675-0.
- A.S. Knoop, H. Knudsen, E. Balslev, B.B. Rasmussen, J. Overgaard, K.V. Nielsen, A. Schonau, K. Gunnarsdóttir, K.E. Olsen, H. Mouridsen, and B. Ejlertsen, Retrospective analysis of topoisomerase IIa amplifications and deletions as predictive markers in primary breast cancer patients randomly assigned to cyclophosphamide, methotrexate, and fluorouracil or cyclophosphamide, epirubicin, and fluorouracil Danish Breast Cancer Cooperative Group, J. Clin. Oncol. 23 (2005), pp. 7483–7490. Erratum in: J. Clin. Oncol. 24 (2006), pp. 1015. doi:10.1200/JCO.2005.11.007.
- C.H. Liang, L.Y. Shiu, L.C. Chang, H.M. Sheu, and K.W. Kuo, Solagine upregulation of Fas, downregulation of HER2, and enhancement of cytotoxicity using epirubicin in NSCLC cells, Mol. Nutr. Food Res. 51 (2007), pp. 999–1005. doi:10.1002/mnfr.200700044.
- F.A. Fornari, J.K. Randolph, J.C. Yalowich, M.K. Ritke, and D.A. Gewirtz, Interference by doxorubicin with DNA unwinding in MCF-7 breast tumor cells, Mol. Pharmacol. 45 (1994), pp. 649–656.
- R.L. Momparler, M. Karon, S.E. Siegel, and F. Avila, Effect of adriamycin on DNA, RNA, and protein synthesis in cell-free systems and intact cells, Cancer Res. 36 (1976), pp. 2891–2895.
- C.A. Frederick, L.D. Williams, G. Ughetto, G.A. van der El, J.H. van Boom, A. Rich, and A.H. Wang, Structural comparison of anticancer drug-DNA complexes: Adriamycin and daunomycin, Biochemistry 29 (1990), pp. 2538–2549. doi:10.1021/bi00462a016.
- X. Chen, Z.L. Ji, and Y.Z. Chen, TTD: Therapeutic target database, Nucleic Acids Res. 30 (2002), pp. 412–415. doi:10.1093/nar/30.1.412.
- A. Rody, T. Karn, C. Solbach, R. Gaetje, M. Munnes, S. Kissler, E. Ruckhäberle, G.V. Minckwitz, S. Loibl, U. Holtrich, and M. Kaufmann, The erbB2+ cluster of the intrinsic gene set predicts tumor response of breast cancer patients receiving neoadjuvant chemotherapy with docetaxel, doxorubicin and cyclophosphamide within the GEPARTRIO trial, Breast 16 (2007), pp. 235–240. doi:10.1016/j.breast.2007.02.006.
- H. Koehn, N. Magan, R.J. Isaacs, and K.M. Stowell, Differential regulation of DNA repair protein Rad51 in human tumour cell lines exposed to doxorubicin, Anticancer Drugs 18 (2007), pp. 419–425. doi:10.1097/CAD.0b013e328012a9a0.
- G. Aubel-Sadron and D. Londos-Gagliardi, Daunorubicin and doxorubicin, anthracycline antibiotics, a physicochemical and biological review, Biochimie 66 (1984), pp. 333–352. doi:10.1016/0300-9084(84)90018-x.
- F. Zunino and G. Canico, DNA topoisomerase II as the primary target of anti-tumor anthracyclines, Anticancer Drug Des. 5 (1990), pp. 307–317.
- Z. Hajihassan and A. Rabbani-Chadegani, Studies on the binding affinity of anticancer drug mitoxantrone to chromatin, DNA and histone proteins, J. Biomed. Sci. 16 (2009), pp. 31. doi:10.1186/1423-0127-16-31.
- J. Mazerski, S. Telli, and E. Borowski, The geometry of intercalation complex of antitumor mitoxantrone and ametantrone with DNA: Molecular dynamics simulations, Acta. Biochim. Pol. 45 (1998), pp. 1–11.
- K. Takeda, K. Shinohara, N. Kameda, and K. Ariyoshi, A case of therapy-related acute myeloblastic leukemia with t(16;21)(q24;q22) after chemotherapy with DNA-topoisomerase II inhibitors, etoposide and mitoxantrone, and the alkylating agent, cyclophosphamide, Int. J. Hematol. 67 (1998), pp. 179–186. doi:10.1016/s0925-5710(97)00108-4.
- H. Wang, Y. Mao, N. Zhou, T. Hu, T.S. Hsieh, and L.F. Liu, ATP-bound topoisomerase II as a target for antitumor Drugs, J. Biol. Chem. 276 (2001), pp. 15990–15995. doi:10.1074/jbc.M011143200.
- Z. Zahraei and A. Rabbani-Chadegani, A comparison of the effect of anticancer drugs, idarubicin and adriamycin, on soluble chromatin, Eur. J. Pharmacol. 575 (2007), pp. 28–33. doi:10.1016/j.ejphar.2007.07.045.
- L.M. Hollingshead and D. Faulds, Idarubicin: A Review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in the chemotherapy of cancer, Drugs 42 (1991), pp. 690–719. doi:10.2165/00003495-199142040-00010.
- T. Fukushima, T. Ueda, M. Uchida, and T. Nakamura, Action mechanism of idarubicin (4-demethoxydaunorubicin) as compared with daunorubicin in leukemic cells, Int. J. Hematol. 57 (1993), pp. 121–130.
- E. Willmore, F. Errington, M.J. Tilby, and C.A. Austin, Formation and longevity of idarubicin-induced DNA topoisomerase II cleavable complexes in K562 human leukaemia cells, Biochem. Pharmacol. 63 (2002), pp. 18007–18015. doi:10.1016/s0006-2952(02)00920-6.
- R. Zhou, Y. Wang, A. Gruber, R. Larsson, E. Castaños-Vèlez, and E. Liliek, Topoisomerase II-mediated alterations of K562 drug resistant sublines, Med. Oncol. 16 (1999), pp. 191–198. doi:10.1007/BF02906131.
- National Center for Biotechnology Information. PubChem compound summary for CID 72092, Otilonium Bromide, PubChem (2020). Available at https://pubchem.ncbi.nlm.nih.gov/compound/Otilonium-Bromide.
- A.J. Sucher, E.B. Chahine, and H.E. Balcer, Echinocandins: The newest class of antifungals, Ann. Pharmacother. 43 (2009), pp. 1647–1657. doi:10.1345/aph.1M237.
- V.A. Morrison, Caspofungin: An overview, Expert Rev. Anti-Infect. Ther. 3 (2005), pp. 697–705. doi:10.1586/14787210.3.5.697.
- P.L. McCormack and C.M. Perry, Caspofungin: A review of its use in the treatment of fungal infections, Drugs 65 (2005), pp. 2049–2068. doi:10.2165/00003495-200565140-00009.
- V. Ries, R. Selzer, T. Eichhorn, W.H. Oertel, K. Eggert, and G. Tolcapone, Replacing a dopamine agonist by the COMT-inhibitor tolcapone as an adjunct to L-dopa in the treatment of Parkinson’s disease: A randomized, multicenter, open-label, parallel-group study, Clin. Neuropharmacol. 33 (2010), pp. 142–150. doi:10.1016/j.bbrc.2007.02.156.
- D.R. Guay, Tolcapone, a selective catechol-o-methyltransferase inhibitor for treatment of Parkinson’s disease, Pharmacotherapy 19 (1999), pp. 6–20. doi:10.1592/phco.19.1.6.30516.
- T.J. Abrams, L.B. Lee, L.J. Murray, N.K. Pryer, and J.M. Cherrington, SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer, Mol. Cancer Ther. 2 (2003), pp. 471–478.
- D.B. Mendel, A.D. Laird, X. Xin, S.G. Louie, J.G. Christensen, G. Li, R.E. Schreck, T.J. Abrams, T.J. Ngai, L.B. Lee, L.J. Murray, J. Carver, E. Chan, K.G. Moss, J.O. Haznedar, J. Sukbuntherng, R.A. Blake, L. Sun, C. Tang, T. Miller, S. Shirazian, G. McMahon, and J.M. Cherrington, In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamics relationship, Clin. Cancer Res. 9 (2003), pp. 327–337.
- A.M. O’Farrell, J.M. Foran, W. Fiedler, H. Serve, R.L. Paquette, M.A. Cooper, H.A. Yuen, S.G. Louie, H. Kim, S. Nicholas, M.C. Heinrich, W.E. Berdel, C. Bello, M. Jacobs, P. Scigalla, W.C. Manning, S. Kelsey, and J.M. Cherrington, An innovative phase I clinical study demonstrates inhibition of FLT3 phosphorylation by SU11248 in acute myeloid leukemia patients, Cancer Res. 9 (2003), pp. 5465–5476.
- R. Roskoski, Sunitinib: A VEGF and PDGF receptor protein kinase and angiogenesis inhibitor, Biochem. Biophys. Res. Commun. 356 (2007), pp. 323–328. doi:10.1016/j.bbrc.2007.02.156.
- S.E. Deprimo, C.L. Bello, J. Smeraglia, C.M. Baum, D. Spinella, B.I. Rini, M.D. Michaelson, and R.J. Motzer, Circulating protein biomarkers of pharmacodynamic activity of sunitinib in patients with metastatic renal cell carcinoma: Modulation of VEGF and VEGF-related proteins, J. Transl. Med. 5 (2007), pp. 1–11. doi:10.1186/1479-5876-5-32.
- J. Guo, P.A. Cotte, J.O. McCall, Y. Dai, L.J. Pease, M.R. Michaelides, S.K. Davidsen, and K.B. Glaser, Inhibition of phosphorylation of the colony-stimulating factor-1 receptor (c-Fms) tyrosine kinase in transfected cells by ABT-869 and other tyrosine kinase inhibitors, Mol. Cancer Ther. 5 (2006), pp. 1007–1013. doi:10.1158/1535-7163.MCT-05-0359.
- H. Prenen, J. Cools, N. Mentens, C. Folens, R. Sciot, P. Schöffski, A. Van Oosterom, P. Ynen, and M. Debiec-Rychter, Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate, Clin. Cancer Res. 12 (2006), pp. 2622–2627. doi:10.1517/14656566.2013.783019.
- K. Kobayashi, S. Yamagami, T. Higuchi, M. Hosokawa, and K. Chiba, Key structural features of ligands for activation of human pregnane X receptor, Drug Metab. Dispos. 32 (2004), pp. 468–472. doi:10.1124/dmd.32.4.468.
- C. Gruber and D. Gruber, Bazedoxifene (Wyeth), Curr. Opin. Investig. Drugs 5 (2004), pp. 1086–1093.
- B.S. Komm, Y.P. Kharode, P.V.N. Bodine, H.A. Harris, C.P. Miller, and C.R. Lyttle, Bazedoxifene acetate: A selective estrogen receptor modulator with improved selectivity, Endocrinology 146 (2005), pp. 3999–4008. doi:10.1210/en.2005-0030.
- National Center for Biotechnology Information. PubChem Compound Summary for CID 10074640, Masitinib, PubChem (2020). Available at https://pubchem.ncbi.nlm.nih.gov/compound/Masitinib.
- F. Cervantes and A. Tinez-Trillos, Myelofibrosis: An update on current pharmacotherapy and future directions, Expert Opin. Pharmacother. 14 (2013), pp. 873–884. doi:10.1517/14656566.2013.783019.
- L.P.H. Yang and G.M. Keating, Ruxolitinib: In the treatment of myelofibrosis, Drugs 72 (2012), pp. 2117–2127. doi:10.2165/11209340-000000000-00000.
- H.U. Bryant, A.L. Glasebrook, N.N. Yang, and M. Sato, An estrogen receptor basis for raloxifene action in bone, J. Steroid Biochem. Mol. Biol. 69 (1999), pp. 37–44. doi:10.1016/s0960-0760(98)00147-2.
- A.J. Krieg, S.A. Krieg, B.S. Ahn, and D.J. Shapiro, Interplay between estrogen response element sequence and ligands controls in vivo binding of estrogen receptor to regulated genes, J. Biol. Chem. 279 (2004), pp. 5025–5034. doi:10.1074/jbc.M307076200.
- H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, and P.E. Bourne, The protein data bank, Nucleic Acids Res. 28 (2000), pp. 235–242. doi:10.1093/nar/28.1.235.
- W.R. Shadrick, J. Ndjomou, R. Kolli, S. Mukherjee, A.M. Hanson, and D.N. Frick, Discovering new medicines targeting helicases: Challenges and recent progress, J. Biomol. Screen. 18 (2013), pp. 761–781. doi:10.1177/1087057113482586.
- S. Charak, D.K. Gir, G. Tyagi, and R. Mehrotra, Interaction studies of epirubicin with DNA using spectroscopic techniques, J. Mol. Struct. 1000 (2011), pp. 150–154. doi:10.1016/j.molstruc.2011.06.013.
- S.R. Byrn and G.D. Dolch, Analysis of binding of daunorubicin and doxorubicin to DNA using computerized curve‐fitting procedures, J. Pharm. Sci. 67 (1978), pp. 688–693. doi:10.1002/jps.2600670532.
- C. Ozluer and H.E.S. Kara, In vitro DNA binding studies of anticancer drug idarubicin using spectroscopic techniques, J. Photochem. Photobiol. B 138 (2014), pp. 36–42. doi:10.1016/j.jphotobiol.2014.05.015.