
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Many cholesterol-laden foam cells in atherosclerotic lesions are macrophages and much of their cholesterol is present in their lysosomes and derived from low density lipoprotein (LDL). LDL oxidation has been proposed to be involved in the pathogenesis of atherosclerosis. We have shown previously that LDL can be oxidised in the lysosomes of macrophages. α-Tocopherol has been shown to inhibit LDL oxidation in vitro, but did not protect against cardiovascular disease in large clinical trials. We have therefore investigated the effect of α-tocopherol on LDL oxidation at lysosomal pH (about pH 4.5). LDL was enriched with α-tocopherol by incubating human plasma with α-tocopherol followed by LDL isolation by ultracentrifugation. The α-tocopherol content of LDL was increased from 14.4 ± 0.2 to 24.3 ± 0.3 nmol/mg protein. LDL oxidation was assessed by measuring the formation of conjugated dienes at 234 nm and oxidised lipids (cholesteryl linoleate hydroperoxide and 7-ketocholesterol) by HPLC. As expected, LDL enriched with α-tocopherol was oxidised more slowly than control LDL by Cu2+ at pH 7.4, but was not protected against oxidation by Cu2+ or Fe3+ or a low concentration of Fe2+ at pH 4.5 (it was sometimes oxidised faster by α-tocopherol with Cu2+ or Fe3+ at pH 4.5). α-Tocopherol-enriched LDL reduced Cu2+ and Fe3+ into the more pro-oxidant Cu+ and Fe2+ faster than did control LDL at pH 4.5. These findings might help to explain why the large clinical trials of α-tocopherol did not protect against cardiovascular disease.
Introduction
Atherosclerosis is a chronic inflammatory disease of the large and medium-sized arteries and the underlying cause of coronary heart disease and thrombotic strokes, which are major causes of morbidity and mortality worldwide [Citation1]. Much of the cholesterol that accumulates in atherosclerotic lesions is contained within foam cells, many of which are derived from macrophages and within macrophages much of the cholesterol is contained within lysosomes [Citation2,Citation3]. There has been much interest in the oxidation of low density lipoprotein (LDL) as one of the key mechanisms of atherogenesis [Citation4,Citation5]. Vitamin E (mainly α-tocopherol) is the major lipid-soluble antioxidant in the body and α-tocopherol is the major antioxidant in LDL with an average of 6 molecules per LDL particle [Citation6]. Epidemiological studies have suggested that dietary antioxidants, including vitamin E, might diminish the risk of coronary heart disease [Citation7] and many experimental studies, almost all of which have been conducted at pH 7.4, have shown that α-tocopherol inhibits LDL oxidation in vitro [Citation8,Citation9]. α-Tocopherol might also affect atherosclerosis by non-antioxidant mechanisms [Citation10]. The large clinical trials, however, failed to show that vitamin E decreases cardiovascular disease [Citation11–20].
We have shown previously that LDL can be oxidised by iron in the lysosomes of macrophages [Citation21] and that this causes the secretion of inflammatory cytokines [Citation22]. The antioxidant cysteamine, which accumulates in lysosomes, inhibits the lysosomal oxidation of LDL [Citation22–25] and decreases atherosclerosis in LDL receptor-deficient mice [Citation25]. The oxidation of LDL by ferrous ion at lysosomal pH (pH 4.5) was not effectively inhibited by α-tocopherol [Citation26]. We have now investigated in more detail the effect of α-tocopherol on LDL oxidation by iron or copper at lysosomal pH.
Material and methods
LDL Isolation and enrichment with α-tocopherol
LDL (d = 1.019–1.063 g/mL) was isolated by sequential ultracentrifugation of pooled plasma from three or two healthy adult volunteers [Citation27]. LDL was enriched with α-tocopherol by incubating plasma at 37 °C for 3 h with α-tocopherol (1 mM) dissolved in dimethyl sulfoxide (1% v/v of the plasma volume) or with dimethyl sulfoxide alone (1% of the plasma volume) as a control [Citation28]. LDL was then isolated by sequential ultracentrifugation, followed by dialysis against 140 mM NaCl/8.1 mM Na2HPO4/1.9 mM NaH2PO4/100 µM EDTA, pH 7.4 [Citation27] during which the dimethyl sulfoxide would have been removed. The α-tocopherol content of the LDL was assessed by extraction with methanol and hexane [Citation29] and reverse phase HPLC [Citation26].
Measurement of conjugated dienes
The oxidation of polyunsaturated lipids in the LDL particles causes the formation of lipid conjugated dienes, which contain two conjugated double bonds. Conjugated dienes can be measured directly using a spectrophotometer as they absorb UV strongly at 234 nm [Citation30]. Control LDL and α-tocopherol-enriched LDL (50 µg LDL protein/mL) was oxidised by freshly disolved FeSO4, FeCl3 or CuSO4 at 37 °C in Chelex-100-treated 150 mM NaCl/10 mM sodium acetate buffer, pH 4.5 or with CuSO4 in Chelex-100-treated 150 mM NaCl/10 mM 3-(N-morpholino)propanesulfonic acid (MOPS) buffer, pH7.4, in quartz cuvettes. Extra equimolar FeSO4, FeCl3 or CuSO4 was added to allow for the EDTA carried over from the stored LDL preparation, which contained 100 µM EDTA to minimise its oxidation during storage. The increase in attenuance at 234 nm was measured every minute at 37° C against reference cuvettes containing all the components except LDL in a double beam Lambda Bio 40 or Lambda Bio 35 8-cell spectrophotometer with UV WinLab software (PerkinElmer). The data were transferred to Excel and attenuance at time zero subtracted from all the time points. The term attenuance was used when LDL oxidation was measured at acidic pH as this refers to both absorbance and scattering of UV, as LDL undergoes aggregation at acidic pH and scatters UV.
It was important to wash the cuvettes well between experiments to avoid contaminating transition ions being present. The cuvettes were rinsed with water several times and washed in warm dilute washing-up liquid using cotton buds. They were then rinsed with purified water, soaked in absolute ethanol for 15 min, rinse with purified water and filled with 100 µM diethylenetriaminepentaacetic acid and left for 1 h. The cuvettes were rinsed with purified water, rinsed with absolute ethanol and left to dry upside down.
HPLC analysis of LDL
The oxidised lipids and α-tocopherol of the LDL were quantified by reverse-phase HPLC as described previously [Citation26] using a HPLC PerkinElmer Series 200 or an Agilent 1100 instrument.
Monitoring of cuprous copper levels
Cuprous ion quantification was achieved by the chelator bathocuproinedisulfonic acid [Citation31]. LDL (50 µg protein/mL) was oxidised with freshly dissolved CuSO4 (5 µM in 150 mM NaCl/10 mM sodium acetate buffer, pH 4.5) in 15 mL polypropylene tubes in a water bath at 37 °C. Samples of 1 mL were taken at various times to new tubes, bathocuproinedisulfonic acid (3 µL of 100 mM) was added and absorbance was measured immediately at 480 nm with a spectrophotometer
Monitoring of ferrous iron levels
The ferrous iron chelator bathophenanthrolinedisulfonic acid was used to measure ferrous iron (Fe2+) levels [Citation32]. LDL (50 µg protein/mL) was oxidised with freshly dissolved FeCl3 (5 µM in 150 mM NaCl/10 mM sodium acetate buffer, pH 4.5) in 15 mL polypropylene tubes in a water bath at 37 °C. Samples of 1 mL were taken at several time points to new tubes, bathophenanthrolinedisulfonic acid (3 µL of 100 mM) was added and absorbance was measured immediately at 535 nm with a spectrophotometer.
Results
Effect of α-tocopherol on LDL oxidation induced by cupric ions at pH 7.4
We enriched LDL with α-tocopherol by incubating plasma with α-tocopherol disolved in dimethyl sulfoxide and then isolating LDL [Citation28]. (Adding α-tocopherol to LDL in a simple buffer does not lead to its incorporation into the LDL particles [Citation28].) The α-tocopherol content of LDL was increased from 14.4 ± 0.2 to 24.3 ± 0.3 nmol/mg protein (mean ± SEM of 5 independent experiments). This corresponds to an average of 7.4 to 12.5 molecules of α-tocopherol per LDL particle, respectively. The fold increase of α-tocopherol of 1.69 corresponds to the fold increase of plasma α-tocopherol obtained in the clinical trials of α-tocopherol and cardiovascular disease (1.60–2.42) [Citation11,Citation14–16,Citation18].
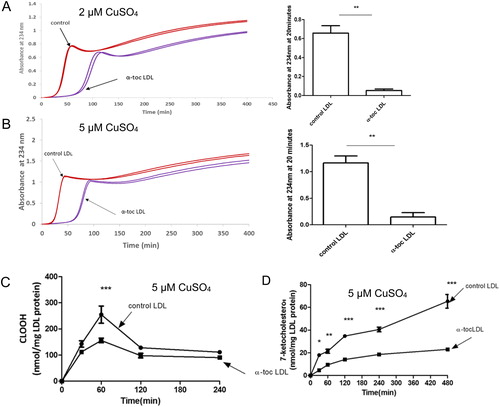
Numerous studies have used copper ions at pH 7.4 to oxidise LDL. We used different concentrations of copper, as α-tocopherol has a greater pro-oxidant effect at lower metal concentrations [Citation33]. At both copper concentrations (2 and 5 µM), α-tocopherol enrichment of LDL delayed the oxidation of LDL, in terms of the formation of conjugated dienes, as expected (). We measured the levels of cholesteryl linoleate hydroperoxide () and 7-ketocholesterol () in the LDL by HPLC. The cholesteryl linoleate hydroperoxide increased faster than the 7-ketocholesterol, as it is oxidised in its polyunsaturated fatty acid moiety, whereas the cholesterol molecule has only one double bond. The levels of cholesteryl linoleate hydroperoxide peaked and then declined because it is converted to more highly oxidised molecules. The levels of cholesteryl linoleate hydroperoxide and 7-ketocholesterol were decreased significantly by α-tocopherol enrichment of LDL.
Figure 1. Oxidation of control LDL and LDL enriched with α-tocopherol by copper at pH 7.4. LDL (50 µg LDL protein/mL) was incubated with (A) 2 or (B) 5 µM CuSO4 at pH 7.4 and 37 °C. Oxidation was monitored by measuring the change in absorbance at 234 nm. The term absorbance is used here as LDL does not aggregate and scatter UV during oxidation under these conditions at pH 7.4. The graphs shown are representative examples of 4 independent experiments. The absorbance (mean ± SEM of 4 independent experiments) at 20 min was measured because it corresponds to substantial LDL oxidation but is before the decomposition phase. The levels of (C) cholesteryl linoleate hydroperoxide (CLOOH) and (D) 7-ketocholesterol in the presence of 5 μM CuSO4 were measured in 3 independent experiments. The absorbance values in (A, B) were compared by a t-test of 4 independent experiments. The CLOOH and 7-ketocholesterol levels were compared by a two-way ANOVA followed by a Bonferroni post-hoc test. *p < 0.05, **p < 0.01, ***p < 0.001.
Effect of α-tocopherol on LDL oxidation induced by cupric ions at pH 4.5
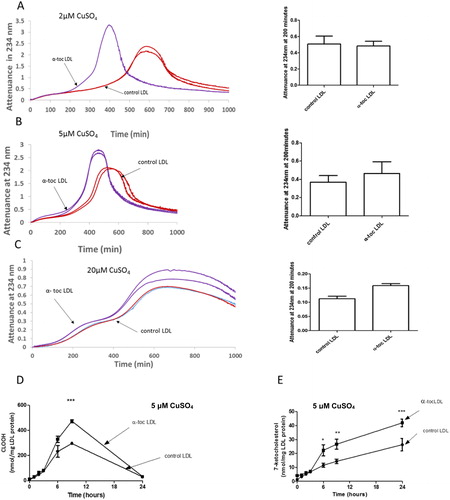
We investigated the effect of α-tocopherol supplementation of LDL on its oxidation by copper at lysosomal pH (pH 4.5) (). LDL oxidation by copper at pH 4.5 and 7.4 are very different. The oxidation is much slower at pH 4.5 than at pH 7.4 (in contrast to ferrous ion where it is much faster at pH 4.5 than pH 7.4 [Citation21,Citation26]). At pH 4.5, it had a short lag phase, a rapid oxidation phase, a slow oxidation phase, an aggregation phase (as seen by an increase in attenuance due to UV scattering by the aggregated LDL particles) and a sedimentation phase as the aggregates fell below the beam of UV in the spectrophotometer resulting in an apparent fall in attenuance. Increasing the concentration of Cu2+ did not greatly affect the rate of oxidation of LDL. (Increasing the concentration of the higher valency state of iron, ferric iron (Fe3+), in contrast to ferrous iron (Fe2+), also does not increase the rate of oxidation of LDL at pH 4.5 [Citation26].) α-Tocopherol supplementation of LDL did not inhibit the oxidation of LDL by any concentration of copper at pH 4.5 (), in marked contrast to the antioxidant effect seen at pH 7.4. α-Tocopherol supplementation of LDL significantly increased the rate of formation of cholesteryl linoleate hydroperoxide () and 7-ketocholesterol () by 5 µM CuSO4 at pH 4.5.
Figure 2. Oxidation of control LDL and LDL enriched with α-tocopherol by copper at pH 4.5. LDL (50 µg LDL protein/mL) was incubated with (A) 2, (B) 5 or (C) 20 µM CuSO4 at pH 4.5 and 37 °C. Oxidation was monitored by measuring the change in attenuance at 234 nm. The graphs shown are representative examples of 4 independent experiments. The attenuance at 200 min (mean ± SEM of 4 independent experiments) was measured because substantial oxidation had occurred by this time, but it had not yet entered the aggregation phase. The levels of cholesteryl linoleate hydroperoxide and 7-ketocholesterol in the presence of 5 µM CuSO4 were measured by HPLC (mean ± SEM of 3 independent experiments). *p < 0.05, **p < 0.01, ***p < 0.001 compared to the control LDL by two-way ANOVA followed by a Bonferroni post-hoc test.
Effect of α-tocopherol on LDL oxidation by ferric ion at pH 4.5
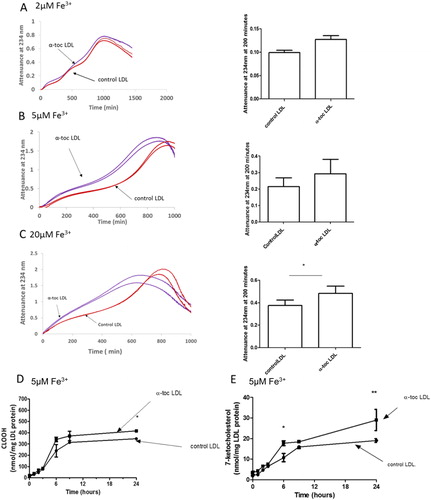
Lysosomes might contain both ferric (Fe3+) and ferrous (Fe2+) iron [Citation34] and we therefore measured the effect of α-tocopherol on the oxidation of LDL by both ferric and ferrous iron. α-Tocopherol did not affect the oxidation of LDL by 2 µM, 5 µM or 20 µM Fe3+ at pH 4.5 (). There was a modest, but statistically significant, pro-oxidant effect of α-tocopherol at 20 µM Fe3+. Supplementation of LDL with α-tocopherol modestly increased the formation of CLOOH () and 7-ketocholesterol () by 5 µM Fe3+ at pH 4.5.
Figure 3. Oxidation of control LDL and LDL enriched with α-tocopherol by ferric iron at pH 4.5. LDL (50 µg LDL protein/mL) was incubated with (A) 2, (B) 5 or (C) 20 µM FeCl3 at pH 4.5 and 37 °C. Oxidation was monitored by measuring the change in attenuance at 234 nm. The graphs shown are representative examples of three independent experiments. The attenuance (mean ± SEM of three independent experiments) at 200 min was measured because substantial oxidation had occurred by this time, but it had not yet entered the aggregation phase. The levels of (D) cholesteryl linoleate hydroperoxide (CLOOH) and (E) 7-ketocholesterol in the presence of 5 μM FeCl3 were measured by HPLC (mean ± SEM of 3 independent experiments). *p < 0.05, **p < 0.01 compared to the control LDL by two-way ANOVA followed by a Bonferroni post-hoc test.
We could not compare the effects of α-tocopherol supplementation of LDL on its oxidation by iron at pH 4.5 and 7.4, as we did for copper, because iron does not oxidise LDL at pH 7.4 in a simple buffer [Citation21,Citation26].
Effect of α-tocopherol on LDL oxidation by ferrous ion at pH 4.5
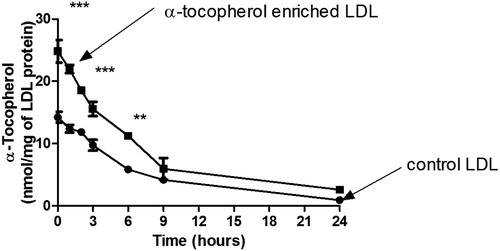
α-Tocopherol supplementation of LDL inhibited LDL oxidation significantly by 5 or 20 µM Fe2+, but not significantly by 2 µM Fe2+ at pH 4.5 (). α-Tocopherol supplementation of LDL decreased the rate of formation of cholesteryl linoleate hydroperoxide () and 7-ketocholesterol () during oxidation by 5 µM Fe2+. The consumption of α-tocopherol in LDL in the presence of 5 µM FeSO4 at pH 4.5 was measured (). The levels of α-tocopherol were increased by supplementation. The levels of α-tocopherol in the control LDL and α-tocopherol-enriched LDL decreased steadily during incubation with Fe2+ until very little was present at 24 h.
Figure 4. Oxidation of control LDL and LDL enriched with α-tocopherol by ferrous iron at pH 4.5. LDL (50 µg LDL protein/mL) was incubated with (A) 2, (B) 5 or (C) 20 µM FeSO4 at pH 4.5 and 37 °C. Oxidation was monitored by measuring the change in attenuance at 234 nm. The graphs shown are representative examples of four independent experiments. The attenuance (mean ± SEM of four independent experiments) at 200 min was measured because substantial oxidation had occurred by this time, but it had not yet entered the aggregation phase. The levels of (D) cholesteryl linoleate hydroperoxide (CLOOH) and (E) 7-ketocholesterol in the presence of 5 μM FeSO4 were measured by HPLC (mean ± SEM of 3 independent experiments). *p < 0.05, **p < 0.01, ***p < 0.001 compared to the control LDL by two-way ANOVA followed by a Bonferroni post-hoc test.
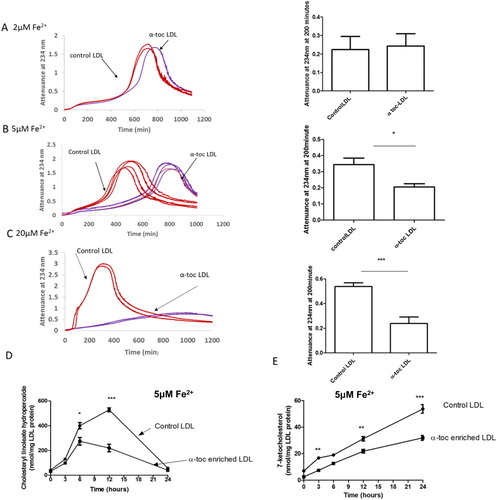
Figure 5. α-Tocopherol consumption during LDL oxidation by ferrous iron at pH 4.5. Control LDL and LDL enriched with α-tocopherol (50 µg LDL protein/mL) was incubated with 5 µM FeSO4 at pH 4.5 and 37 °C and the levels of α-tocopherol were measured by HPLC (mean ± SEM of 3 independent experiments). **p < 0.01, ***p < 0.001 compared to the control LDL by two-way ANOVA followed by a Bonferroni post-hoc test.
Measurement of Cu+ and Fe2+
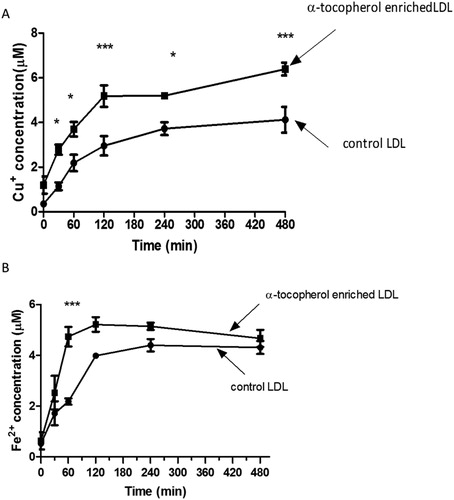
To explore the mechanisms of the antioxidant/pro-oxidant effects of α-tocopherol, we measured the rate of reduction of Cu2+ to Cu+ and Fe3+ to Fe2+ at lysosomal pH (). The reduction was almost complete by 2 h, which corresponds with the rapid oxidation phase of LDL (). α-Tocopherol supplementation of LDL increased the rate of reduction of both metals.
Figure 6. Kinetics of cupric and ferric ion reduction by LDL at pH 4.5. Control and LDL enriched with α-tocopherol (50 μg protein/mL) were incubated with (A) 5 μM CuSO4 or (B) 5 μM FeCl3 at pH 4.5 and 37 °C. At several time points, samples were taken and Cu+ or Fe2+ ion detected immediately using the cuprous copper chelator bathocuproinedisulfonic acid or bathophenanthrolinedisulfonic acid, respectively. The mean ± SEM of 4 independent experiments is shown. *p < 0.05, ***p < 0.001 by two-way ANOVA followed by a Bonferroni post-hoc test.
Discussion
Clinical trials of antioxidants, mainly α-tocopherol, for treatment of cardiovascular disease were conducted because α-tocopherol inhibits LDL oxidation in vitro, but the large trials did not show any protection [Citation11,Citation12,Citation14–19]. This has led some to question the importance of oxidised LDL in atherosclerosis. It has been argued that multiple antioxidants might be required for a disease in which multiple oxidants might be involved or that the trials were conducted with older patients in whom oxidative stress had become progressively less important with age [Citation35,Citation36]. The studies of α-tocopherol and LDL oxidation in vitro were carried out at pH 7.4, however, whereas it is possible that LDL oxidation in atherosclerotic lesions occurs in the lysosomes of macrophages at a pH of about 4.5 catalysed by iron [Citation21,Citation25]. We show here that in marked contrast to the expected inhibition by α-tocopherol of LDL oxidation by Cu2+ at pH 7.4, α-tocopherol does not inhibit LDL oxidation by Cu2+ or Fe3+ at pH 4.5, although it inhibits LDL oxidation by higher concentrations of Fe2+ at pH 4.5. We oxidised LDL with copper, as well as with iron. Iron does not oxidise LDL at pH 7.4 in a simple buffer [Citation21], probably because hydroperoxyl radicals are not formed at this pH [Citation23], and we therefore could not investigate the effect of α-tocopherol on LDL oxidation by iron at pH 7.4. Copper oxidises LDL at both pH 7.4 and 4.5, however, and this enabled us to compare the effects of pH on the antioxidant and pro-oxidant effects of α-tocopherol.
LDL oxidation by copper at pH 7.4 () was much faster than at pH 4.5 (), whereas it is much faster by iron at pH 4.5 than at 7.4 [Citation21]. The mechanism of LDL oxidation by copper at pH 7.4 is controversial, but probably proceeds by a different mechanism to that at pH 4.5. It might involve the interaction of copper with certain amino acids in the protein moiety of LDL, apolipoprotein B-100, such as tryptophan residues [Citation23,Citation37], which we hypothesise might occur less at acidic pH.
Copper was added in the cupric form (Cu2+), but cycles between the cupric and cuprous (Cu+) forms during its catalysis of LDL oxidation. Cu+ would react with oxygen to form the superoxide radical (O2•–), which would protonate at acidic pH to form the hydroperoxyl radical (HO2•), which has a pKa of 4.8. Very little protonation would occur at pH 7.4. As discussed before [Citation23,Citation38], the hydroperoxyl radical is much more reactive than the superoxide radical and, unlike the superoxide radical, is not charged and therefore has access to the oxidisable bisallylic methylene groups of polyunsaturated lipids (LH) of LDL.
LDL oxidised by copper at pH 4.5 aggregated, whereas it did not aggregate at pH 7.4. This might be explained at least in part because at pH 7.4 the LDL particles have a strong net negative charge, which would repel the LDL particles from each other, whereas at pH 4.5 they would have a weaker net positive charge [Citation26].
α-Tocopherol usually inhibits LDL oxidation at pH 7.4, as shown by numerous studies [Citation8,Citation9]. This is probably due mainly to α-tocopherol (α-tocOH) scavenging lipid peroxyl radicals [Citation33].
α-Tocopherol can have a pro-oxidant effect, however, especially if the oxidative stress is low, as shown by Stocker’s group [Citation33,Citation39], a process termed tocopherol-mediated peroxidation. α-Tocopherol can react with cupric (Cu2+) [Citation40] or ferric (Fe3+) ions to produce two pro-oxidants: (1) the α-tocopheroxyl radical and (2) the cuprous (Cu+) or ferrous (Fe2+) ion.
The α-tocopheroxyl radical would eventually abstract a hydrogen atom from a polyunsaturated fatty acyl moiety causing lipid peroxidation [Citation33,Citation39].
The cuprous ion or ferrous ion would react with lipid hydroperoxides (LOOH) much faster than would cupric ion or ferric ion (Fe3+) [Citation6].
In support of this, we showed that α-tocopherol supplementation of LDL increased the rate of conversion of Cu2+ to Cu+ and of Fe3+ to Fe2+ (). (It should be noted that the Cu+ and Fe2+ indicators, bathocuproinedisulfonic acid and bathophenanthrolinedisulfonic acid, respectively, were not present during the incubation of the LDL with Cu2+ and Fe3+ and therefore would not have artefactually pulled the reaction over in the direction of Cu+ and Fe2+.)
α-Tocopherol supplementation of LDL (by incubating plasma with α-tocopherol and isolating the LDL by ultracentrifugation) strongly decreased the rate of LDL oxidation by copper ions at pH 7.4, as expected (). Remarkably, α-tocopherol had no antioxidant effect and might even have had a pro-oxidant effect with copper ions at pH 4.5 (). These different effects might be explained by the net balance of the antioxidant and pro-oxidant reactions of α-tocopherol being altered by a change in pH.
We postulate that α-tocopherol was less effective at inhibiting LDL oxidation at acidic pH because the hydroperoxyl radicals present at acidic pH might abstract the phenolic hydrogen atom from α-tocopherol forming the α-tocopheroxyl radical.
The α-tocopheroxyl radical is not entirely stable and would eventually abstract a hydrogen atom from a polyunsaturated fatty acyl moiety causing lipid peroxidation [Citation33,Citation39], as described above. The estimated rate constant for the reaction of α-tocopherol and hydroperoxyl radicals is 1.5 × 105 M−1 s−1 [Citation41], which we expect would be greater than that for the reaction of hydroperoxyl radicals with the bisallylic methylene group of polyunsaturated lipids.
The α-tocopherol might in effect “trap” the lipid oxidising power of hydroperoxyl radicals inside the LDL particles (as α-tocopheroxyl radicals) causing more oxidation of the lipids in LDL.
Another possible reason to explain why α-tocopherol inhibited LDL oxidation at pH 7.4 but not at pH 4.5, is as follows. At high oxidative stress there might be an α-tocopheroxyl radical and another free radical present in a single LDL particle at the same time which might react with each other to from a nonradical product, decreasing the pro-oxidant effect of α-tocopherol [Citation33].
The oxidation of LDL by copper ions was much faster at pH 7.4 than 4.5, which might have decreased the pro-oxidant effect of α-tocopherol by increasing the probability of there being two free radicals in a single LDL particle at one time. This might have contributed to a net antioxidant effect of α-tocopherol prevailing with copper at pH 7.4.
α-Tocopherol enrichment of LDL did not inhibit LDL oxidation by the higher valence state of iron, ferric ion (Fe3+) at pH 4.5 (), similarly to the lack of antioxidant effect with the higher valence state of copper, cupric ion (Cu2+) at pH 4.5. There was a statistically significant, but modest, pro-oxidant effect with 20 µM Fe3+ and sometimes with 5 μM Fe3+.
Supplementation with α-tocopherol delayed the oxidation of LDL by 5 or 20 µM ferrous ions (Fe2+) (), in contrast to the lack of antioxidant effect observed with ferric ion. Ferrous ion, in contrast to ferric ion, would not have directly converted α-tocopherol into the potentially pro-oxidant α-tocopheroxyl radical. Also, the greater antioxidant effect of α-tocopherol with ferrous ions than ferric ions might possibly be due in part to the faster rate of oxidation of LDL by ferrous () than ferric ions (), as there would have been a greater possibility of having two radicals in a LDL particle at the same time with a faster rate of oxidation, lessening the pro-oxidant effect of α-tocopherol, as discussed above. In support of this, α-tocopherol had an antioxidant effect with ferrous ion at higher oxidative stress (5 or 20 µM Fe2+), but not at lower oxidative stress (2 µM Fe2+) ().
In a previous study [Citation26], we showed that supplementation of LDL with α-tocopherol caused an immediate pro-oxidant effect with 5 µM ferrous ion at pH 4.5 followed by an antioxidant effect. The reason for the difference in these results from our present results is not clear, but might possibly be due to the level of enrichment obtained with α-tocopherol.
The enrichment in α-tocopherol we obtained in LDL (1.69 fold) was comparable to that achieved in the clinical trials of α-tocopherol and cardiovascular disease (1.60 − 2.42) [Citation11,Citation14–16,Citation18]. The lack of inhibition of LDL oxidation due to α-tocopherol enrichment by Cu2+ or Fe3+ (or sometimes a pro-oxidant effect) or a low concentration of Fe2+ at pH 4.5 might help to explain why the large clinical trials of α-tocopherol were ineffective. It would be desirable to conduct clinical trials of antioxidants that accumulate in lysosomes and inhibit the oxidation of LDL at acidic pH, such as cysteamine [Citation22–25].
Author contributions
H.K.M.A. carried out the experiments and H.K.M.A. and D.S.L. wrote the paper.
Acknowledgements
We thank the volunteers who donated their blood for LDL isolation and Dr Kim G. Jackson and Ms Rada Mihaylova of Food and Nutritional Sciences, University of Reading for skilfully taking it. H.K.M.A. was supported by the University of Karbala, Iraq.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Rafieian-Kopaei M, Setorki M, Doudi M, et al. Atherosclerosis: process, indicators, risk factors and new hopes. Int J Prev Med. 2014;5(8):927–946.
- Shio H, Haley NJ, Fowler S. Characterization of lipid-laden aortic cells from cholesterol-fed rabbits. II. Morphometric analysis of lipid-filled lysosomes and lipid droplets in aortic cell populations. Lab Invest. 1978;39(4):390–397.
- Peters TJ, de Duve C. Lysosomes of the arterial wall. II. subcellular fractionation of aortic cells from rabbits with experimental atheroma. Exp Mol Pathol. 1974;20(2):228–256.
- Chisolm GM, Steinberg D. The oxidative modification hypothesis of atherogenesis: an overview. Free Radic Biol Med. 2000;28(12):1815–1826.
- Tsimikas S, Miller YI. Oxidative modification of lipoproteins: mechanisms, role in inflammation and potential clinical applications in cardiovascular disease. Curr Pharm Des. 2011;17(1):27–37.
- Esterbauer H, Gebicki J, Puhl H, et al. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic Biol Med. 1992;13(4):341–390.
- Gey KF, Puska P. Plasma vitamins E and A inversely correlated to mortality from ischemic heart disease in cross-cultural epidemiology. Ann NY Acad Sci. 1989;570(1 Vitamin E):268–282.
- Jessup W, Rankin SM, de Whalley CV, et al. a-Tocopherol consumption during low-density-lipoprotein oxidation. Biochem J. 1990;265(2):399–405..
- Dieber-Rotheneder M, Puhl H, Waeg G, et al. Effect of oral supplementation with D-a-tocopherol on the vitamin E content of human low density lipoproteins and resistance to oxidation. J Lipid Res. 1991;32:1325–1332.
- Azzi A, Stocker A. Vitamin E: non-antioxidant roles. Prog Lipid Res. 2000;39(3):231–255.
- Collins R, Armitage J, Parish S, et al. MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:23–33.
- Yusuf S, Phil D, Dagenais G, et al. Vitamin E supplementation and cardiovascular events in high-risk patients. N Engl J Med. 2000;342(3):154–160.
- Brown BG, Cheung MC, Lee AC, et al. Antioxidant vitamins and lipid therapy: end of a long romance? Arterioscler Thromb Vasc Biol. 2002;22(10):1535–1546.
- Cook NR, Albert CM, Gaziano JM, et al. A randomized factorial trial of vitamins C and E and beta carotene in the secondary prevention of cardiovascular events in women – Results from the women’s antioxidant cardiovascular study. Arch Intern Med. 2007;167(15):1610–1618.
- Hodis HN, Mack WJ, LaBree L, et al. Alpha-tocopherol supplementation in healthy individuals reduces low-density lipoprotein oxidation but not atherosclerosis: the Vitamin E Atherosclerosis Prevention Study (VEAPS). Circulation. 2002;106(12):1453–1459.
- Keith ME, Jeejeebhoy KN, Langer A, et al. A controlled clinical trial of vitamin E supplementation in patients with congestive heart failure. Am J Clin Nutr. 2001;73(2):219–224.
- Lee IM, Cook NR, Gaziano JM, et al. Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women’s Health Study: a randomized controlled trial. J Am Med Assoc. 2005;294(1):56–65.
- Lonn E, Bosch J, Yusuf S, et al. Effects of long-term vitamin E supplementation on cardiovascular events and cancer – a randomized controlled trial. J Am Med Assoc. 2005;293:1338–1347.
- Sesso HD, Buring JE, Christen WG, et al. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians’ Health Study II randomized controlled trial. J Am Med Assoc. 2008;300(18):2123–2133.
- Vivekananthan DP, Penn MS, Sapp SK, et al. Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet. 2003;361(9374):2017–2023.
- Wen Y, Leake DS. Low density lipoprotein undergoes oxidation within lysosomes in cells. Circ Res. 2007;100(9):1337–1343.
- Ahmad F, Leake DS. Lysosomal oxidation of LDL alters lysosomal pH, induces senescence, and increases secretion of pro-inflammatory cytokines in human macrophages. J Lipid Res. 2019;60(1):98–110.
- Ahmad F, Leake DS. Antioxidants inhibit low density lipoprotein oxidation less at lysosomal pH: a possible explanation as to why the clinical trials of antioxidants might have failed. Chem Phys Lipids. 2018;213:13–24.
- Ojo OO, Leake DS. Low density lipoprotein oxidation by ferritin at lysosomal pH. Chem Phys Lipids. 2018;217:51–57.
- Wen Y, Ahmad F, Mohri Z, et al. Cysteamine inhibits lysosomal oxidation of low density lipoprotein in human macrophages and reduces atherosclerosis in mice. Atherosclerosis. 2019;291:9–18.
- Satchell L, Leake DS. Oxidation of low-density lipoprotein by iron at lysosomal pH: implications for atherosclerosis. Biochemistry. 2012;51(18):3767–3775.
- Wilkins GM, Leake DS. The effect of inhibitors of free radical generating-enzymes on low-density lipoprotein oxidation by macrophages. Biochim Biophys Acta. 1994;1211(1):69–78.
- Esterbauer H, Dieber-Rotheneder M, Striegl G, et al. Role of vitamin E in preventing the oxidation of low-density lipoprotein. Am J Clin Nutr. 1991;53:S 314–S 321.
- Kritharides L, Jessup W, Gifford J, et al. A method for defining the stages of low-density lipoprotein oxidation by the separation of cholesterol- and cholesteryl ester-oxidation products using HPLC. Anal Biochem. 1993;213(1):79–89.
- Esterbauer H, Striegl G, Puhl H, et al. Continuous monitoring of in vitro oxidation of human low density lipoprotein. Free Radic Res Commun. 1989;6(1):67–75.
- Perugini C, Seccia M, Albano E, et al. The dynamic reduction of Cu(II) to Cu(I) and not Cu(I) availability is a sufficient trigger for low density lipoprotein oxidation. Biochim Biophys Acta. 1997;1347(2–3):191–198.
- Pieroni L, Khalil L, Charlotte F, et al. Comparison of bathophenanthroline sulfonate and ferene as chromogens in colorimetric measurement of low hepatic iron concentration. Clin Chem. 2001;47(11):2059–2061.
- Thomas SR, Stocker R. Molecular action of vitamin E in lipoprotein oxidation: implications for atherosclerosis. Free Radic Biol Med. 2000;28(12):1795–1805.
- Meguro R, Asano Y, Odagiri S, et al. The presence of ferric and ferrous iron in the nonheme iron store of resident macrophages in different tissues and organs: histochemical demonstrations by the perfusion-Perls and -Turnbull methods in the rat. Arch Histol Cytol. 2005;68(3):171–183.
- Niki E. Oxidant-specific biomarkers of oxidative stress. Association with atherosclerosis and implication for antioxidant effects. Free Radic. Free Radic Biol Med. 2018;120:425–440.
- Steinberg D, Witztum JL. Is the oxidative modification hypothesis relevant to human atherosclerosis? Do the antioxidant trials conducted to date refute the hypothesis? Circulation. 2002;105(17):2107–2111.
- Gieβauf A, Steiner E, Esterbauer H. Early destruction of tryptophan residues of apolipoprotein B is a vitamin E-independent process during copper-mediated oxidation of LDL. Biochim Biophys Acta-Lipids Lipid Metab. 1995;1256(2):221–232.
- Bedwell S, Dean RT, Jessup W. The action of defined oxygen-centred free radicals on human low-density lipoprotein. Biochem J. 1989;262(3):707–712.
- Bowry VW, Ingold KU, Stocker R. Vitamin E in human low-density lipoprotein – When and how this antioxidant becomes a prooxidant. Biochem J. 1992;288(2):341–344.
- Yoshida Y, Tsuchiya J, Niki E. Interaction of alpha-tocopherol with copper and its effect on lipid peroxidation. Biochim Biophys Acta. 1994;1200(2):85–92..
- Navarrete M, Rangel C, Espinosa-Garcia J, et al. Theoretical study of the antioxidant activity of vitamin E: Reactions of α-Tocopherol with the Hydroperoxy Radical. J Chem Theory Comput. 2005;1(2):337–344.