Abstract
A multiparticulate system combining pH-sensitive property and specific biodegradability for colon-targeted delivery of metronidazole has been investigated. Cross-linked chitosan microspheres were prepared from an emulsion system using liquid paraffin as the external phase and solution of chitosan in acetic acid as the disperse phase. The multiparticulate system was prepared by coating cross-linked chitosan microspheres exploiting Eudragit® L-100 and S-100 as pH-sensitive polymers. Morphology and surface characteristics of the formulations were determined by scanning electron microscopy. Particle size of the chitosan microspheres was determined by optical microscopy while that of coated microspheres was determined by particle size analyzer. In vitro drug-release studies were performed in conditions simulating stomach-to-colon transit in presence and absence of rat caecal contents. The size of the microspheres was small and they were efficiently microencapsulated within Eudragit® microspheres, forming a multireservoir system. By coating the microspheres with Eudragit® pH-dependant release profiles were obtained. No release was observed at acidic pH; however, when it reached the pH where Eudragit® starts solublizing there was continuous release of drug from the formulation. Further, the release of drug was found to be higher in the presence of rat caecal contents, indicating the susceptibility of chitosan matrix to colonic enzymes released from rat caecal contents.
INTRODUCTION
Targeting of drugs specifically to colon is advantageous in the treatment of diseases associated with the colon such as amebiasis, Crohn's diseases, ulcerative colitis, and colorectal cancer. In addition, it has shown great potential in oral delivery of therapeutic peptides and proteins, which are unstable in the upper part of gastrointestinal tract. The colonic region is recognized as having less diversity and intensity of enzymatic activities than the stomach and small intestine (Davis Citation1990). Various strategies are available for targeting drug release selectively to the colon (Chourasia and Jain Citation2003). The designing of prodrugs is based on the concept of preventing the release of drugs in the stomach and small intestine, and drug release is triggered by the utilization of some specific property at the target site such as altered pH or high activity of certain enzymes in comparison to nontarget tissues (Davaran et al. Citation1999; Schacht et al. Citation1996). Since it is known that azo function can be reduced in the colon (Chung et al. Citation1992), a lot of novel polymers containing azo groups either in the polymeric backbone (Yamaoka et al. Citation2000) or in the crosslinks (Shantha et al. Citation1995; Van den Mooter et al. Citation1992) have been synthesized. In order to promote further selective degradation in the vicinity of colonic environment, delivery systems have been designed that contain both pH-sensitive acidic monomers and degradable azo aromatic crosslinks (Chandehari et al. Citation1997; Kakoulides et al. Citation1998). Polysaccharides such as chitosan, dextran, inulin, and guar gum have been explored for their potential in colon-specific drug delivery (Prasad et al. Citation1998; Shimono et al. Citation2002; Tozaki et al. Citation2002). The polysaccharides remain intact in the hostile environment of the stomach and small intestine, and upon arrival in the colon they are degraded by polysaccharidases (Rubinstein et al. Citation1993).
pH-sensitive polymers, which dissolve at or above pH 7, may also be used for colonic delivery (Cole et al. Citation2002; Khan et al. Citation1999). Ashford et al. (Citation1993) showed that pH-sensitive polymers are not suitable for colon-targeted drug delivery systems due to poor site specificity. The long lag time at the ileocaecal junction and fast transit indicate that a single unit may not be the best dosage form for colon-targeted drug delivery system.
The proposed multiparticulate system combines pH-sensitive property of enteric polymers as well as biodegradability of chitosan in the colon. It consists of chitosan microspheres coated using pH-sensitive polymers for the colon-targeted delivery of metronidazole (MNZ) for the treatment of amebiasis. The drug release is suppose to take place after dissolution of the enteric coating in the small intestine and biodegradation of the chitosan in the colon due to presence of polysaccharidases in the colonic contents.
MATERIALS AND METHODS
Chitosan (purified viscosity grade 50) was obtained from Central Institute of Fisheries Technology (Cochin, India). M/s Broshell Pharmaceuticals (Sagar, M. P., India) generously supplied metronidazole as a gift sample. Span-80 and Antifoam A were procured from Sigma Chemicals (St.Louis, MO, USA). Liquid paraffin heavy (viscosity 90 cp at 30°) was from Central Drug House (Mumbai, India). All other solvents and reagents were of analytical grade.
Preparation of Cross-Linked Chitosan Microspheres
The microspheres were prepared using the emulsion method, employing glutaraldehyde as cross-linker (Thanoo et al. Citation1992). Chitosan solution (4% w/v) was prepared in 5% aqueous acetic acid, and the drug was dispersed in this solution and mixed well. This was dispersed in liquid paraffin (1:1 mixture of light and heavy) containing span 80 (1% w/w) and antifoam A (0.1% w/w). The dispersion was stirred using a stainless steel half moon paddle stirrer at various speeds for 2 min and glutaraldehyde-saturated toluene solution (1 ml to 3 ml) was added under stirring and continued for 4 hr. After the stipulated stirring time, the microspheres were centrifuged, washed several times with hexane, and dried in vacuum desiccator for 48 hr.
Coating of Cross-Linked Chitosan Microspheres
Coating of cross-linked chitosan microspheres containing MNZ was performed using emulsion solvent evaporation technique. Chitosan microspheres were suspended in 10 ml of an organic solvent (1:1, acetone:methanol) in which Eudragit ® L-100 or S-100 was previously dissolved to give either 1:5 or 1:10 core/coating ratio. This organic phase was emulsified into 100 ml of liquid paraffin containing span 80 and antifoam A (1 and 0.1% w/v, respectively). The system was stirred at 1000 rpm with two-blade mechanical stirrer for 4 hr at room temperature. The Eudragit®-coated microspheres were collected and rinsed with n-hexane and dried in vacuum desiccator for 48 hr.
In Vitro Characterization of Microspheres
Determination of Particle Size and Shape
Morphology and surface characteristics of the microspheres were determined using scanning electron microscopy (AIIMS, New Delhi, India).
Particle size of the cross-linked chitosan microspheres was determined by optical microscopy using calibrated ocular eyepiece, whereas that of Eudragit®-coated microspheres was determined using particle size analyzer (Cilas 1064 L, Marcoussis, France).
Determination of Drug Content
The amount of drug present in the microspheres was determined by a method reported by Thanoo et al. (Citation1992). A weighed quantity of the microspheres was extracted with methanol for 24 hr, and drug concentration in supernatant was determined spectrophotometrically at 320.5 nm (UV 1601, Shimadzu, Japan).
In Vitro Drug Release from Microspheres
In vitro drug release studies were carried out according to Souder and Ellenbogen (Citation1985) extraction technique using USP dissolution test apparatus. The dissolution studies were carried out in 100 ml dissolution medium, which was stirred at 100 rpm at 37± 0.1°C.
The scheme of using the simulated fluids at different pH was as follows:
1st hour: Simulated gastric fluid of pH 1.2.
2nd and 3rd hours: Mixture of simulated gastric and intestinal fluid of pH 4.5.
4th and 5th hours: Simulated intestinal fluid of pH 6.8.
6th hour: Simulated intestinal fluid of pH 7.5.
In Vitro Drug Release in Presence of Rat Caecal Contents
In vitro drug release from coated microspheres was also carried out in the presence of rat caecal contents to assess the biodegradability of chitosan by colonic bacteria. Albino rats of either sex weighing between 150–200 g were selected for the present study and maintained on normal diet. In order to induce the enzymes that specifically act on the chitosan during its passage through the colon, the albino rats were intubated with Teflon tubing and 1 ml of 1% w/v dispersion of chitosan in water was administered directly into the stomach. This treatment was continued for five days. Rats were dissected before release rate studies, the caecum was isolated, ligated at both ends, cut loose and immediately transferred into simulated intestinal fluid of pH 7.5 previously bubbled with carbon dioxide. The caecal contents were individually weighed, pooled, and suspended in buffer to produce final caecal concentration of 3% w/v. Drug release rate studies for the initial 5 hr were performed as described above. From 6th hour and onwards it was carried out in simulated intestinal fluid containing rat caecal contents. The experiment was carried out with a continuous supply of carbon dioxide into dissolution media. Aliquots of samples were withdrawn periodically and replaced with fresh buffer bubbled with carbon dioxide. The volume was made up to 10 ml, centrifuged, the supernatant was filtered through Whatman filter paper, and drug content was determined spectrophotometrically at 320.5 nm (UV 1601, Shimadzu, Japan).
RESULTS AND DISCUSSION
The multiparticulate systems for colon-targeted drug delivery exhibit an advantage over single-unit dosage form, as they remain intact in the stomach and small intestine. Once they reach in the colonic region, release of the drug is triggered due to degradation of the system. Single unit dosage forms are based on pH-sensitive polymers, which are not suitable for colon-targeted drug delivery. These dosage forms exploit the enteric polymers and maintain their integrity and do not cause the release of the drug in the strongly acidic environment of the stomach. As they arrive into the alkaline pH of the small intestine they start to dissolve and release the drug.
This multiparticulate system combines a pH-sensitive property and biodegradability in the colon. Colon-specific release of the entrapped drug is achieved by dissolution of the enteric coating at the distal part of the small intestine and exposed chitosan microspheres when reaches to the colon, release is effected by swelling of the polymer as well as by the biodegradable effect of polysaccharidases.
When aqueous glutaraldehyde is added to the dispersion of chitosan in paraffin oil, an instantaneous reaction occurs and the resultant product does not exhibit good sphericity and surface texture. Therefore, slow and uniform cross-linking of the droplets, particularly at the surface, is desirable to generate microspheres of good sphericity. Hence, glutaraldehyde-saturated toluene was used, which by virtue of its solubility in oil medium would help to uniformly cross-link the surface of droplets.
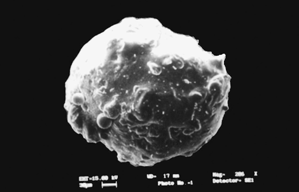
It is clearly seen from scanning electron photomicrograph that microspheres possess a smooth surface and spherical shape (). Particle size and entrapment efficiency of crosslinked chitosan microspheres bearing MNZ is shown in . Due to the strong swelling and adhesive nature of the chitosan, the particle size of cross-linked chitosan microspheres could not be determined by particle size analyzer, and hence calibrated ocular eye piece was used for measurement of particle size by microscopic method. Mean particle size was found to be decreased by increasing the amount of the drug while it was increased on increasing the chitosan concentration. Mean particle size was found to be 14.5± 0.52 μm at 10%, drug concentration, while it was decreased to 12.1± 0.49 μm when the drug concentration was increased to 20%. Increasing the concentration of chitosan caused the viscosity to increase and led to an increase in the size of emulsion droplet, and hence microspheres with bigger particles were produced. Particle size was decreased (14.3± 0.55 to 10.2± 0.21 μm) by increasing the agitation speed (1000 to 3000 rpm). When the agitation speed was increased above 3000 rpm the mean particle size was reduced, and microspheres did not exhibit good sphericity and surface appearance.
1 Scanning electron photomicrograph of cross-linked chitosan microspheres.
Average particle size, entrapment efficiency, and in vitro drug release of uncoated cross-linked chitosan microspheres
The in vitro drug release studies were performed using gastrointestinal fluids of different pH. The effect of drug concentration, chitosan concentration, glutaraldehyde concentration, and agitation speed was observed on in vitro drug release. As the amount of drug incorporated into cross-linked chitosan microspheres was increased, the in vitro release was increased. In vitro drug release after 4 hr was found to be 75.2± 3.41% in the case of microspheres having 10% drug, while it was 84.2± 3.89% for microspheres with 20% drug (). The effect of chitosan concentration on the release of drug was found to be meager (). The drug release after 8 hr was decreased (99.5± 3.94 to 91.4± 3.45%) when the concentration of glutaraldehyde, was increased (1 ml to 3 ml), which is due to the fact that as the amount of glutaraldehyde was increased it produced microspheres with pronounced cross-linking between polymer chains that retarded the release of drug (). Microspheres, which were prepared at agitation speed of 3000 rpm, did not exhibit good sphericity and released 99.4± 3.82% of MNZ, while those that were produced at 2000 rpm released 99.0 ± 3.68% of MNZ after 8 hr (). All the formulations released more than 90% of the drug during dissolution studies for 8 hr.
2 Effect of drug concentration on drug release from cross-linked chitosan microspheres.
In the second part of this investigation cross-linked chitosan microspheres were further coated with Eudragit® L-100 and Eudragit® S-100 using esmulsion solvent evaporation technique.
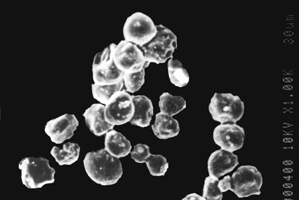
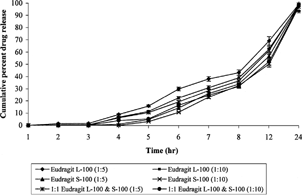
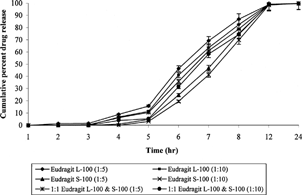
The shape of the Eudragit®-coated cross-linked chitosan microspheres was determined using scanning electron microscopy, and particle size was determined using laser diffraction particle size analyzer (Cilas 1064 L, France). The size after coating was considerably increased (140–175 μm) ( and ). In the case of uncoated cross-linked chitosan microspheres, nearly 80–90% of the drug was released in the initial 4–5 hr release rate studies. This situation is not acceptable for those drugs, which are required to be released locally in the colon. Cross-linked chitosan microspheres were coated with Eudragit® L-100, S-100, and a 1:1 mixture of both polymers to retard the release of the drug until pH reaches above 6.0. In vitro release studies revealed that no drug was released during first 3 hr studies except in case of Eudragit® L-100 (core/coating ratio, 1:5), where 1.4± 0.04% of the drug was released. In the case of microspheres prepared from 1:1 mixture of Eudragit® L-100 and S-100 (core/coating ratio, 1:5), 0.5± 0.02% of the drug was released during the initial 3 hr. Such release could be due to the entrapped drug nearer to surface that was dissolved and diffused out into the medium after swelling. Eudragit® L-100 dissolves at pH above 6.0, and hence during 4th-hour study at pH 6.8, 8.9± 0.39% and 6.7± 0.25% of the drug was released in the case of 1:5 and 1:10 core/coating ratio, respectively, while no drug was released in case of Eudragit® S-100 microspheres (1:10). Moreover, drug was continuously released when the release studies were carried out above solubility pH of the enteric polymers. Eudragit® L-100 microspheres started releasing drug at lower pH (6.8) than Eudragit® S-100 (7.5), while microspheres consisted of mixture of Eudragit® L-100 and S-100 exhibited intermediate release pattern. The MNZ release from the microspheres was significantly affected by coating ratio (core/coating). The higher amount of drug release was observed with microspheres prepared by 1:5 core/coating ratio, while microspheres having 1:10 core/coating ratio exhibited lower drug release (). Drug release after 12 hr in case of Eudragit® L-100 with core/coating ratio of 1:5 and 1:10 was found to be 69.3± 3.12 and 62.7± 2.83%, respectively, whereas microspheres of Eudragit® S-100 with the same core/coating ratio released 56.9± 2.65% and 50.4± 1.98%, respectively.
Particle size and encapsulation efficiency of coated cross-linked chitosan microspheres
3 Scanning electron photomicrograph of coated cross-linked chitosan microspheres.
4 In vitro release of metronidazole from coated cross-linked chitosan microspheres.
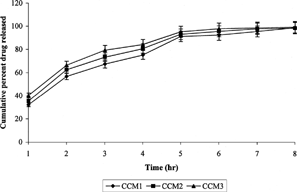
The multiparticulate system successfully retarded the release of drug until it enters into the colon. To assess the biodegradability of the chitosan to the colonic enzymes, in vitro release studies in the presence of caecal contents were performed. The albino rats were orally administered 1 ml of 1% w/v chitosan dispersion in water continuously for 5 days in order to induce the enzymes that specifically act on chitosan. Drug release rate studies in the presence of rat caecal contents were carried out from the 6th hour in simulated intestinal fluid of pH 7.4. Drug release-rate studies for initial 5 hr were the same as described previously.
The presence of rat caecal contents in the dissolution medium resulted in improved drug release at different time intervals in comparison to those that were carried out without using rat caecal contents. In vitro drug release studies after 8 hr without rat caecal contents released 32.8 ± 1.46% and 39.2 ± 1.29% of drug in case of microspheres of 1:10 core/coating of Eudragit® S-100 and L-100, respectively, while the presence of rat caecal contents increased the release to 69.8 ± 3.17% and 82.4 ± 3.28% (). The release of drug from cross-linked chitosan microspheres was supposed to take place after swelling, which resulted in the formation of gel followed by the dissolution of MNZ and diffusion through the gel. Cross-linking of the chitosan with glutaraldehyde caused the swelling to decrease and the consequent release of drug. This particular phenomenon is helpful during the exposure of the chitosan microspheres to small intestinal fluid. Drug release in presence of rat caecal contents is triggered by the swelling behavior of chitosan as well as by biodegradability due to colonic enzymes. More than 98% of MNZ was released from all the formulations after 12 hr studies in presence of rat caecal contents ().
5 In vitro release of metronidazole from coated cross-linked chitosan microspheres in the presence of rat caecal contents.
The release of a higher amount of drug in the presence of rat caecal contents clearly reveals the susceptibility of chitosan matrix to colonic enzymes released from rat caecal contents. A considerably higher amount of caecal matter is present in the human colon than the amount present in the in vitro studies. It may not be necessary to induce the enzymes in case of administration of dosage form of chitosan, as considerable amount of polysaccharidases would be present in the human colon.
The author is grateful to M/s Broshell Pharmaceuticals, Sagar, M. P., India for generously supplying metronidazole as gift sample and Council of Scientific and Industrial Research, New Delhi, India for providing financial assistance to carry out this work.
REFERENCES
- Ashford M., Fell J. T., Attwood D., Sharma H., Woodhead P.. 1993. An in vivo investigation into the suitability of pH-dependent polymers for colonic targeting. Int. J. Pharm.. 95: 193–199
- Chandehari H., Kopeckova P., Kopecek J.. 1997. In vitro degradation of pH sensitive hydrogels containing aromatic azo bonds. Biomaterials. 18: 861–872. [CROSSREF]
- Chourasia M. K., Jain S. K.. 2003. Pharmaceutical approaches to colon targeted drug delivery systems. J. Pharm. Pharmaceut. Sci.. 6(1)33–66
- Chung K. T., Stevens S. E., Cerniglia C. E.. 1992. The reduction of azo dyes by the intestinal microflora. Crit. Rev. Microbiol.. 18: 175–190. [PUBMED], [INFOTRIEVE]
- Cole E. T., Scott R. A., Connor A. L., Wilding I. R., Petereit H. U., Schminke C., Beckert T., Cade D.. 2002. Enteric coated HPMC capsules designed to achieve intestinal targeting. Int. J. Pharm.. 231: 83–95. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Davaran S., Hanaee J., Khosravi A.. 1999. Release of 5-amino salicylic acid from acrylic type polymeric prodrugs designed for colon-specific drug delivery. J. Contrl. Rel.. 58: 279–287. [CROSSREF]
- Davis S. S.. 1990. Overcoming barriers to the oral administration of peptide drugs. Trends Pharma. Sci.. 11: 353–355. [CROSSREF]
- Kakoulides E. P., Smart J. D., Tsibouklis J.. 1998. Azocrosslinked (polyacrylic acid) for colonic delivery and adhesion specificity: in vitro degradation and preliminary ex vivo bioadhesion studies. J. Contrl. Rel.. 54: 95–109. [CROSSREF]
- Khan M. Z., Prebeg Z., Kurjakovic N. A.. 1999. A pH-dependent colon targeted oral drug delivery system using methacrylic acid copolymers I. Manipulation of drug release using Eudragit® L100–55 and Eudragit® S100 combinations. J. Contrl. Rel.. 58: 215–222. [CROSSREF]
- Prasad Y. V. R., Krishnaiah Y. S. R., Satyanarayana S.. 1998. In vitro evaluation of guar gum as a carrier for colon-specific drug delivery. J. Control. Rel.. 51: 281–287. [CROSSREF]
- Rubinstein A., Radai R., Ezra M., Pathak S., Rokem J. M.. 1993. In vitro evaluation of calcium pectinate: A potential colon-specific drug delivery carrier. Pharm. Res.. 10: 258–263. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Schacht E., Gevaert A., Kenawy E. R., Molly K., Gelan T.. 1996. Polymers for colon-specific drug delivery. J. Contrl. Rel.. 39: 327–338. [CROSSREF]
- Shantha K. L., Ravichandran P., Rao K. P.. 1995. Azo polymeric hydrogels for colon targeted drug delivery. Biomaterials. 16: 1313–1318. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Shimono N., Takatori T., Masumi T., Ueda M., Mori M., Higashi Y., Nakamura Y.. 2002. Chitosan dispersed system for colon-specific drug delivery. Int. J. Pharm.. 245: 45–54. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Souder J. C., Ellenbogen W. C.. 1985. Control of d-amphetamine sulphate sustained release capsule. Drug Standards. 26: 77–79
- Thanoo B. C., Sunny M. C., Jayakrishnan A.. 1992. Cross linked chitosan microspheres: Preparation and evaluation as a matrix for the controlled release of pharmaceuticals. J. Pharm. Pharmacol.. 44: 283–286. [PUBMED], [INFOTRIEVE]
- Tozaki H., Odoriba T., Okada N., Fujita T., Terabe A., Suzuki T., Okabe S., Murnishi S., Yamamoto A.. 2002. Chitosan capsules for colon-specific drug delivery: Enhanced localization of 5-aminosalicylic acid in the large intestine accelerates healing of TNBS-induced colitis in rats. J. Contrl. Rel.. 82: 51–61. [CROSSREF]
- Van den Mooter G., Samyn C., Kinget R.. 1992. Azo polymers for colon-specific drug delivery. Int. J. Pharm.. 87: 37–46. [CROSSREF]
- Yamaoka T., Makita Y., Sasatani H., Kim I., Kimura Y.. 2000. Linear type azo-conataining polyeurathane as drug coating material for colon-specific delivery. J. Contrl. Rel.. 66: 187–197. [CROSSREF]