
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Amphotericin B (AMB) is used most commonly in severe systemic life-threatening fungal infections. There is currently an unmet need for an efficacious (AMB) formulation amenable to oral administration with better bioavailability and lower nephrotoxicity. Novel PEGylated polylactic-polyglycolic acid copolymer (PLGA-PEG) nanoparticles (NPs) formulations of AMB were therefore studied for their ability to kill Candida albicans (C. albicans). The antifungal activity of AMB formulations was assessed in C. albicans. Its bioavalability was investigated in nine groups of rats (n = 6). Toxicity was examined by an in vitro blood hemolysis assay, and in vivo nephrotoxicity after single and multiple dosing for a week by blood urea nitrogen (BUN) and plasma creatinine (PCr) measurements. The MIC of AMB loaded to PLGA-PEG NPs against C. albicans was reduced two to threefold compared with free AMB. Novel oral AMB delivery loaded to PLGA-PEG NPs was markedly systemically available compared to Fungizone® in rats. The addition of 2% of GA to the AMB formulation significantly (p < 0.05) improved the bioavailability from 1.5 to 10.5% and the relative bioavailability was > 790% that of Fungizone®. The novel AMB formulations showed minimal toxicity and better efficacy compared to Fungizone®. No nephrotoxicity in rats was detected after a week of multiple dosing of AMB NPs based on BUN and PCr, which remained at normal levels. An oral delivery system of AMB-loaded to PLGA-PEG NPs with better efficacy and minimal toxicity was formulated. The addition of glycyrrhizic acid (GA) to AMB NPs formulation resulted in a significant oral absorption and improved bioavailability in rats.
Introduction
Amphotericin B (AMB) has been the gold standard treatment for severe systemic life-threatening fungal infections since 1959 (Bekersky et al., Citation1999; Cifani et al., Citation2012). It has been used as the first-line treatment for visceral leishmaniasis, a life-threatening parasitic disease, in the endemic area of Bihar, India (Sundar et al., Citation2010). Unfortunately, AMB formulations are only available for parenteral administration.
The amphipathic nature of AMB significantly reduces its solubility in water and most organic solvents. Its aqueous solubility is improved by adding sodium deoxycholate to produce a colloidal dispersion after reconstitution for intermittent intravenous (iv) infusion (Fungizone®). However, severe side effects are associated with the administration of Fungizone®. Nephrotoxicity is the most serious chronic adverse effect of AMB; the serum creatinine concentration (Scr) increases in > 80% of patients receiving the drug (Sachs-Barrable et al., Citation2008; Tonomura et al., Citation2009; Chuealee et al., Citation2011). Additionally, AMB could induce hematological side-effects (Brajtburg & Bolard, Citation1996; Yu et al., Citation1998; Adams & Kwon, Citation2003).
There is limited information regarding AMB metabolism and tissue distribution (Egger et al., Citation2001). The primary route of its elimination is not known (Drew, Citation2013). AMB state (monomers or aggregates) affects its efficacy and toxicity. Nishi and his coinvestigators have suggested that AMB is therapeutically active in its monomeric forms while the existence of aggregates forms are responsible for its toxicity (Nishi et al., Citation2007).
The development of parenteral AMB lipid-based formulations, Abelcet®, Ambisome® and Amphocil®, which have shorter course of therapy (3–5 days), are effective and exhibit lower toxicity when compared to Fungizone® (Torrado et al., Citation2008; Sundar et al., Citation2010). However, their cost has restricted widespread use (Sachs-Barrable et al., Citation2008; Falamarzian & Lavasanifar, Citation2010).
AMB is also characterized by instability at gastric pH and is unable to cross the mucosal barrier of the GI tract and enter the blood stream. There has been some effort to formulate AMB for oral administration. These include formulating AMB as nanosuspensions (Kayser et al., Citation2003), as Poly(lactide-co-glycolide) nanoparticles (NPs) employing vitamin E-TPGS as a stabilizer (Italia et al., Citation2009; Italia et al., Citation2011), as lipid-based oral formulation using Peceol (Sachs-Barrable et al., Citation2008) or as liquid antisolvent precipitation NPs (Zu et al., Citation2014). Furthermore, AMB has been loaded to Peceol and PEG-phospholipids (iCo-009) (Gershkovich et al., Citation2010; Sivak et al., Citation2011), to carbon nanotubes (Prajapati et al., Citation2012), to gelatin-coated lipid NPs (Jain et al., Citation2012), to Chitosan–EDTA conjugates (Singh et al., Citation2013) and to Cubosomes (Yang et al., Citation2012; Yang et al., Citation2014). The most recent reports include AMB liposomes containing ceramides (Skiba-Lahiani et al., Citation2015) and AMB encapsulated with a chitosan derivative (Serrano et al., Citation2015). These oral drug deliveries were developed to enhance the solubility and gastrointestinal permeability of AMB. In most cases, these formulations failed to increase the absorption of orally administered AMB and none of them has been introduced to the market (Ibrahim et al., Citation2012; Yang et al., Citation2012).
Glycyrrhizic acid (GA) is a major constituent of licorice, a triterpene glycoside, with steroid-like, antiallergic and antiviral activities (Pompei et al., Citation1980). It has been used orally as a sweetener and component of oriental medicines (Imai et al., Citation1999; Anand et al., Citation2010). In the field of drug delivery, GA possesses in vivo enhancing activity with respect to the oral absorption of peptides such as calcitonin (Imai et al., Citation1999). It is reported as non-toxic oral absorption enhancer to improve the oral bioavailability of different drugs (Radwan & Aboul-Enein, Citation2002; Chen et al., Citation2009; Yang et al., Citation2015).
We hypothesized that loading AMB to PEGylated polylactic–polyglycolic acid copolymer (PLGA–PEG) NPs would improve AMB solubility; decrease its toxicity (since the drug release would be controlled from this delivery system) and AMB aggregation (thereby further decreasing its toxicity toward mammalian cells) while maintaining it in a monomeric form that favors antifungal activity (Brajtburg & Bolard, Citation1996; Torrado et al., Citation2008).
Two reports on the use of PLGA–PEG NPs as AMB parenteral delivery systems has recently published. The first study was concern about delivering AMB as a mannose-anchored engineered nanoparticulate for macrophage targeting (Nahar & Jain, Citation2009), while the second paper encapsulated AMB in PLGA-PEG NPs to increase AMB solubility and to target the macrophages of infected tissues during visceral leishmaniasis (Kumar et al., Citation2015). To our knowledge, no published data about the development of AMB loaded to PLGA-PEG NPs for oral AMB delivery other than by Al-Quadeib et al. (Citation2015); the in vitro studies of the developed formulations indicated promising oral drug delivery system with acceptable drug content and dissolution rate. The aim of this study was to examine the practicability, efficacy, oral bioavailability and safety of these novel AMB formulations in rats. The in vitro AMB antifungal activity on Candida albicans (C. albicans) of these formulations was initially evaluated. Pharmacokinetics investigation of formulations after iv and oral administrations to rats and the feasibility of GA as an absorption enhancer to improve AMB bioavailability was then investigated. Toxicity was examined via an in vitro blood hemolysis test and via blood urea nitrogen (BUN) and plasma creatinine (PCr), as indicators of nephrotoxicity after single and multiple dosing of AMB in rats.
Materials and methods
Materials
AMB (99.8% purity), clopidogrel, the internal standard (IS), GA and formic acid were purchased from Sigma–Aldrich (St. Louis, MO).
PEGylated Poly (D,L-lactide-co-glycolide) copolymer: RGPd 50155 Diblock with molecular weight 6000 Da (Lactic to glycolic acid molar ratio of 1:1) with 15% polyethylene glycol and Poly(D,L-lactide): Poly(D,L-lactide) named R 203 H Monoblock were supplied by Boehringer Ingelheim (Ingelheim, Germany). Fungizone® (AMB micelle dispersion) was obtained from Bristol-Myers Squibb (Montreal, Canada). RPMI 1640 medium was purchased from Gibco/BRL (Grand Island, NY). Sabouraud Dextrose Agar plates (RODAC™) were obtained from BD Diagnostics (Becton, Dickinson and Company, NJ). All other reagents and chemicals were of HPLC analytical grade, and were used as received. Water was deionized and purified by a Milli-Q Reagent Grade water system (Millipore Corporation, Bedford, MA).
Preparation of AMB-loaded PLGA-PEG copolymer
AMB loaded to PLGA-PEG was formulated according to Al-Quadeib et al (Citation2015) with modification through addition of GA during formulation or prior to administration as shown below. shows the criteria of the selected AmB NPs, from previous study; having a narrow size distribution with smallest possible mean particle size, highest drug encapsulation efficacy and drug release within 24 h. Physicochemical characterizations (particle size analysis, FTIR, DSC and dissolutions were done in our previously published work (Al-Quadeib et al., Citation2015).
Table 1. The characteristics of the selected formulations.
AMB loaded to PLGA–PEG NPs formulation (C6) was selected as the base formulation for this study and is named F1. When GA was added to F1 just prior to administration the formulation is called F1-GA-out-1 and F1-GA-out-2, where, 1 and 2 refer to the percentage of GA added. When GA 2% (w/v) was added to the organic phase during F1 preparation, the resulted formulation is called F1-GA-in-2.
F2 was prepared by doubling the amount of AMB in the formulation C6. When GA was added during preparation, the formulation is called F2-GA-in-2, while F2-GAout-2 is the same composition but the GA was added just prior to administration. To investigate the feasibility of PLGA-PEG NPs versus non-PEGylated polymer, PLA, AMB loaded to PLA NPs formulation (F3) was prepared by the same technique of C7. All formulations were prepared at least in triplicate.
In vitro antifungal activity
Candida albicans (ATCC 90028) (C. albicans) was used for testing the efficacy of AMB in vitro. The minimum inhibitory concentrations (MICs) of AMB in Fungizone®, F1 and F2 were determined by broth dilution according to the National Committee for Clinical Laboratory Standard “NCCLS document M27-A, (Standards, Citation2002)”. Briefly, C. albicans cell suspensions of ∼1 × 106 cells/ml were diluted 1:50 in RPMI-1640 growth medium and 100 μl dispensed into a microliter tray containing a serial concentration of AMB 0.05–1.5 μg/ml. A solution of 5 mg/ml was prepared in DMSO for free AMB and in water for Fungizone®, F1 and F2 immediately before use. The tray was incubated for 24 and 48 h at 37 °C. The yeast were grown on Sabouraud Dextrose Agar (SDA) plates and inoculated into RPMI 1640 broth medium to yield a final inoculum concentration of 104 yeast cells/ml (checked by doing a viable colony count on SDA plates). Two wells containing drug-free medium and inoculum were used as control. The inoculated plates were incubated at 35 °C for 24 h. The growth in each well was estimated visually. The MIC was defined as the lowest drug concentration (MIC) that resulted in complete inhibition of visible growth. The MIC was recorded to be the lowest concentration of the AMB that prevented visible growth of C. albicans and expressed in μg/ml. The end-point was determined as the concentration to produce optically clear wells (MIC-0).
Dosing of animals and blood sampling
All the experiments were performed in accordance with the ethical guidelines established and approved by the committee on the use and care of laboratory animals at King Saud University. Animals were provided from the animal house of King Saud University. The rats were housed in polypropylene cages in an animal facility with a 12 h light-dark cycle and controlled temperature and humidity. Rats were given free access to water ad libitum and fasten for 10 h before the study. Standard rat chow was given after 2 h of dosing for the duration of the study.
shows the composition and method of in vivo administration of the formulations tested in rats.
Table 2. The composition, dose and route of administration of the selected formulations.
Immediately before administration, the specific weight of the lyophilized AMB-loaded PLGA–PEG NPs were dispersed in water either alone or in combination with GA as 1 or 2% (w/v) as indicated. The volume of the PO colloidal dispersion dose was 1.0 ml.
The study was conducted into two phases; phase I was intended to examine the bioavailability of AMB in the selected formulations while phase II was designed to investigate the toxicity of AMB in these formulations as shown below.
After animals dosing, blood samples (600 μl) were withdrawn from the venous-orbital plexus from each rat in heparinized tubes. Carbon dioxide was used to slightly anesthetize the animals during blood sampling. Plasma samples were separated by centrifugation at 4000 rpm for 15 min and were stored at −20 °C prior to drug assay.
Bioavailability study
Fifty-four albino Sprague-Dawley male rats (353.2 ± 26.6 g) were randomly divided into nine groups (I–IX, n = 6) and were dosed according to . Blood samples from the same rats were collected at 0, 1, 3, 6 and 0.5, 2, 4, 8, 24 h after drug administration in two different occasions, separated by 3 weeks.
Nephrotoxicity and histopathological studies
The selected formulations of AMB loaded to PLGA-PEG NPs for the in vitro efficacy and in vitro/in vivo toxicity investigations were F1 and F2, which contain 20 and 40 mg of AMB, respectively, without any addition of GA.
Fifteen albino Sprague-Dawley rats with 200–300 g weight were randomly divided into five groups (1–5), (n = 3). Group-1 was kept as control without any treatment; group-2 received iv blank NPs with no AMB; group 3 was given Fungizone® (1.0 mg/kg) as a slow iv injection in the tail vein; group-4 received iv administration of F1 (1.0 mg/kg) while group-5 received iv administration of F2 (1.0 mg/kg).
After 24 h post first dose, blood samples were collected from each rat as the single dose samples (SD) experiment. The animals were received daily the same treatment for 7-days and 24 h post-the 7th dose, blood samples were collected for the multiple dose (MD) experiment.
BUN and PCr were measured using an automatic analyzer 7180 (Hitachi High- Technologies Co., Tokyo, Japan) after the single and multiple dosing administrations.
For histopathological analysis, one kidney from each rat was fixed in neutral buffered 10% formalin for ≥48 h, bisected and embedded in paraffin. Sections of 5.0 μm thickness were cut from each kidney, and stained with hematoxylin and eosin. For histopathological analysis of liver, a piece of median lobe from the livers were removed from each rats and placed immediately in 10% neutral buffered formalin. Sections of 5.0 μm thickness were cut and stained with hematoxylin and eosin similarly to kidney tissue.
In vitro hemolytic activity of AMB
In vitro hemolytic activity of AMB-loaded to PLGA-PEG NP of the selected formulations (F1 and F2) were assessed using isolated rat red blood cells (RBCs) according to Jain and Kumar (Jain & Kumar, Citation2010). Briefly, blood samples from healthy Sprague-Dawley male rats (250–350 g) were collected by cardiac puncture under anesthesia directly into heparinized blood collecting vials. The RBCs were separated by centrifuging the whole blood at 3000 rpm for 15 min, the supernatant along with buffy coat were pipetted off and discarded. RBCs were washed thrice with 0.15 M isotonic phosphate buffer saline (PBS) of pH 7.4 and was dispersed in PBS to obtain 1% hematocrit. RBCs were used on the same day for further experiments. Subsequently, 1.0 ml of the RBCs suspension was mixed with 1.0 ml of PBS containing 20, 50 or 100 μg/ml AMB equivalent formulations (Fungizone®, F1 or F2) and was incubated at 37 °C in a shaking water bath at 100 rpm. The experiment was performed in triplicate. After 8 and 24 h of incubation, any hemolysis was stopped by reducing the temperature to 0° C and un-lysed RBCs were removed by centrifugation for 10 min at 3000 rpm. The supernatant was collected and the erythrocyte pellet was lysed with sterile distilled water and analyzed for the extent intact RBCs using a spectrophotometer set at the absorption maximum of hemoglobin (540 nm). Control RBC (2 × 108 cells/ml) incubated with PBS alone was used to estimate the total hemoglobin content.
Incubation of RBCs with distilled water (as positive control) was considered to cause 100% hemolysis. Results were expressed as a percentage of hemolysis as given in the following equation:
Where, AbsS is the absorbance of the sample, Abs0 is the average absorbance of the buffer; negative control, and Abs100 is the average absorbance of the lysed samples (in purified water; positive control). The remaining hemoglobin was calculated as a percentage of the total content. Results are given as the mean of one experiment representative of three experiments carried out with each concentration in triplicate.
Chromatographic conditions
Analysis was carried out on a Waters Acquity UPLC™ system (Waters, Milford, MA). The analytical method used was adapted from Al‐Quadeib et al. (Citation2014) using rat plasma instead of human plasma. There was no significant difference (p > 0.05) between the standard curves best fit equations on slopes, intercepts and correlation of the human and rat plasma. The precision and accuracy of the developed LC MS/MS method were measured for the concentration range of 100–4000 ng/ml and was shown no significant difference among inter- and-intra-day analysis (p > 0.05) in rat plasma. Excellent linearity was observed over the investigated range with correlation coefficient, r > 0.995 (n = 6/day). The assay was able to detect AMB concentrations for all time points after iv and PO administrations to rats without any modification.
Pharmacokinetics and statistical analysis
All data were expressed as the mean ± SD, if not specified otherwise, of six replicates for the assay or the rats study. The standard curves were calculated by linear regression without weighting, using the equation Y = 0.0007X – 0.127, where Y is the AUP ratio of the drug to the IS, −0.127 is the intercept, 0.0007 is the slope, and X is AMB concentration. The relative standard deviation (RSD) was calculated for all values.
The plasma AMB concentration versus time data of the rats were analyzed using a model-independent method for the main purpose of this study (Gibaldi & Perrier, Citation1982). The mean maximum concentration (Cmax) and the time to reach Cmax (Tmax) were derived directly from the individual plasma levels. The elimination rate constant (k) was calculated from the slope of the regression line that best fit the terminal part (last three to four points) of the log–linear concentration–time profile of AMB. The terminal half-life (t1/2) was calculated from 0.693/k. The area under the curve from time 0 to 24 h (AUC0–24) was estimated by linear trapezoidal rule and was extrapolated to time infinity (AUC) by the addition of Cn/k where, Cn is concentration of the last measured plasma sample. The total body clearance (Cl) was estimated using the equation Cl = dose/AUC. The volume of distribution (V) was determined from the equation k = Cl/V. For oral data, the absorption rate constant ka was estimated by the method of residual (Tozer & Rowland, Citation2006). The absolute bioavailability (F) was calculated by the equation (AUCpo/Dosepo)/(AUCiv/Doseiv) after iv of F1 and oral administration of the tested formulations listed in while the relative bioavailability (Frel) was calculated using (AUC, PO formulation/AUC, F1-PO) Frel to Fungizone (AUC, PO formulation/AUC, Fungizone-PO).
All statistical differences in data were evaluated using IBM SPSS Statistics 21 (IBM cooperation, New York, NY). The Student t-test was used to examine the concentration difference at each day and one-way analysis of variance (ANOVA) was employed to assess the reproducibility of the assay and other tests were used when needed. p < 0.05 was considered significant.
Results
Bioavailability of amphotericin B in rats
Novel oral biodegradable stealth polymeric nanoparticles of AMB have been successfully fabricated by a modified emulsification–diffusion technique using PLGA–PEG (Al-Quadeib et al., Citation2015). shows the logarithmic mean plasma concentration versus time profiles of AMB following F1 formulation as a 1.0 mg/kg single dose (F1-iv) and 10 mg/kg (F1-PO) to two different groups of rats via iv and PO administrations, respectively. Fungizone®, 10 mg/kg, (Fungizone®-PO) was given to a third group of rats via PO administration for comparison.
Figure 1. Mean plasma AMB concentration–time profiles after intravenous administration of 1.0 mg/kg of F1-iv and oral administrations of 10 mg/kg of F1-PO and Fungizone® to rats (n = 6).
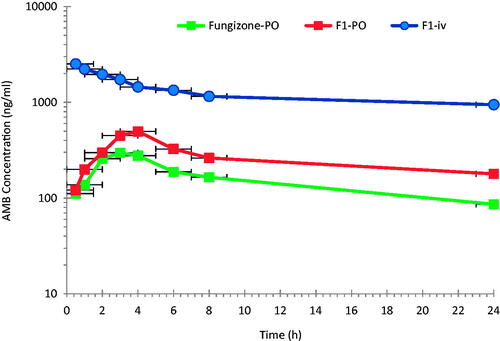
After iv administration, a two compartment open model was considered to adequately describe the kinetics of AMB in rat plasma with a fast distribution, over the first 4 h, followed by slow elimination. The oral administration of AMB as F1-PO or Fungizone® show the same trend, after the absorption phase. However, a model-independent method was used instead for the main purpose of this study and for simplicity (Gibaldi & Perrier, Citation1982).
The pharmacokinetic parameters of the three AMB dosage forms (F1-iv, F1-PO and Fungizone®-PO) in rats are summarized in . After iv administration of F1-iv, AMB shows a relatively long elimination t1/2 ranging from one to two days in rats due to its low Cl (8–23.2 ml/h/kg) and large apparent volume of distribution in the range of 552.7–806.3 ml/kg.
Table 3. Pharmacokinetic parameters of AMB after iv and oral administrations of AMB-loaded PLGA-PEG formulations and fungizone in rats (n = 6).
Although the iv dose was one tenth the PO doses of either F1-PO or Fungizone® PO, F1-iv shows a pronounced higher plasma concentration of AMB (p < 0.05). A statistically significant higher Cmax of AMB was observed following F1-PO than following Fungizone®-PO administrations. The observed mean plasma AUC0–24 h and AUC0–∞ after F1-PO was about 63.7% and 36.4%, respectively, higher than that of Fungizone®-PO. The bioavailability (F) of F1-PO was 36.4% higher than that of Fungizone® PO due to the improvement of AMB release and absorption when loaded to PLGA-PEG but the results were not satisfactory.
To investigate the effect of GA as an absorption enhancer, F1 and F2 were given to rats with GA, added just prior to the administration or were included in the organic phase during AMB loaded to PLGA-PEG NP preparation. AMB mean plasma concentration–time profile after single oral doses of AMB-loaded PLGA–PEG formulations with and without the addition of the absorption enhancer GA are depicted in and the pharmacokinetic parameters are presented in .
Figure 2. Mean plasma AMB concentration–time profiles following single oral administration of 10.0 mg/kg of AMB-loaded PLGA-PEG formulations in rats (n = 6).
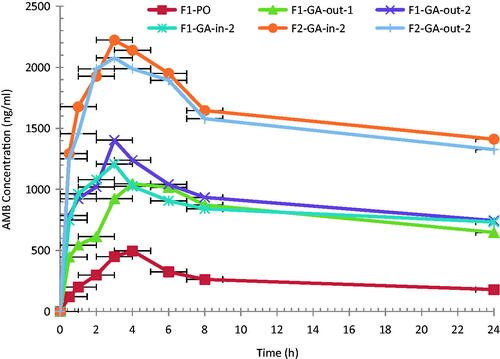
Cmax after F1 without GA was 1.6 times that after Fungizone (p < 0.05). A significant increase (p < 0.05) in AMB Cmax (2.3-fold) with 4.3-fold increase in AUC (332.3%) was observed after the addition of 1% of GA to the PO formulations, just prior to the administration. This is an indication of the efficiency of GA as an absorption enhancer for AMB.
Increasing the GA content from 1% (F1-GA-out-1) to 2% (F1-GA-out-2), showed a significant increase in Cmax (2.97 fold) and in AUC (390.7%) compared to F1 formulation without GA. Further increase in GA content to 3% showed no significant change in either Cmax or AUC of AMB profile, data not shown. Therefore, GA at 2% was selected as the optimum amount to be added to the tested formulations.
It was noticed that the addition of GA during preparation process (F1-GA-in-2) or just prior to the administration (F1-GA-out-2) showed no significant difference (p > 0.05) in the AUC (13.5%), with a 27.9% increase in the Cmax value. This is an indication that the efficacy of GA as an absorption enhancer for AMB is not changed either GA added just prior to administration or during the preparation.
Doubling the amount of the AMB in the formulation from 20 mg (F1) to 40 mg (F2) resulted in doubling the AUC of F1-GA-in-2 and F2-GA-in-2 (20216.2 ± 1593 and 38808.6 ± 4033 μg.h/l, respectively). The same trend was observed when GA was out, therefore, AMB shows linear kinetic within the tested amount in the formulation.
It should be mentioned that for each tested formulation, in rats, the value of the absorption rate constant of AMB loaded to PLGA-PEG NPs, ka, was much greater than the value of elimination rate constant k, which indicates a fast release of AMB with no sign of the flip-flop’ model (Gibaldi & Perrier, Citation1982). The highest ka among the tested formulations was F1-GA-in compare to the others. Therefore, the addition of GA during preparation of F1 resulted in higher release of AMB.
The absolute bioavailability (F) of AMB from tested AMB loaded to PLGA-PEG NP formulation was calculated, . Upon the addition of 2% GA during the formulations, the F was improved from 1.5 to 10.5% (sixfolds). The Frel was improved from 332 to 616.5% and 25 to 800% after the addition of GA compared to F1-PO and Fungizone, respectively. The highest improvement in Frel was detected in formulation F1-GA-in-2.
Table 4. Pharmacokinetic parameters of AMB (mean ± SD) after a 10 mg/kg single oral administration of AMB as Fungizone® and AMB-loaded PLGA-PEG NP formulations in rats (n = 6).
Although the iv dose was one tenth the PO doses of either F1-PO or Fungizone®-PO, F1-iv shows a pronounced higher plasma concentration of AMB (p < 0.05). This could be attributed to either incomplete absorption of AMB from these formulations or because the drug was subjected to a significant first pass metabolism in the liver after absorption from the GI tract.
Moreover, loading AMB to non-PEGlyated polymer (poly (D, L-lactide-co-glycolide, RG 502 H), showed no absorption of AMB at all the time points. This is a confirmation of the importance of the PEG in PLGA polymer to promote the absorption of AMB after PO administration.
In vivo nephrotoxicity
Nephrotoxicity is the most serious adverse effect of AMB after chronic exposure; the Scr increases in more than 80% of patients receiving this drug (Sabra and Branch, Citation1990; Tonomura et al., Citation2009). In the conventional solution formulation, the drug is present as micelles. As soon as the drug solution enters the bloodstream it separates from the micelles and this appears to be critical to subsequent nephrotoxic effects. The incidence of AMB nephrotoxicity is very high, varying in studies between 49% and 65% (Deray, Citation2002). Nephrotoxicity is defined in most studies as a doubling of baseline creatinine (Cr) levels (100% increase from the serum baseline) or greater than 2.5 mg/dl in human (Miller et al., Citation2004).
The present investigation examined two biomarkers (BUN and PCr) of renal injury. No mortality was observed after iv doses in any of the rats during the study. Relevant changes in serum chemistry are summarized in and . In the rats treated with iv Fungizone®, there was a significant (p = 0.003 and p = 0.001) increase in BUN and PCr levels, respectively, as compared to control rats, indicating that Fungizone administration resulted in nephrotoxicity upon administration as a single or multiple doses. However, iv administration of AMB-loaded to PLGA-PEG NPs led to non-significant difference (p > 0.05) in BUN and PCr levels in the rats, as compared to control rats, indicating lower nephrotoxicity by AMB when loaded to PLGA-PEG NPs as compared to Fungizone®.
Figure 3. BUN, mg/dL, (mean ± SD) after single and multiple iv doses (5 mg/kg) of AMB and AMB loaded to PLGA-PEG NP to rats (n = 3).
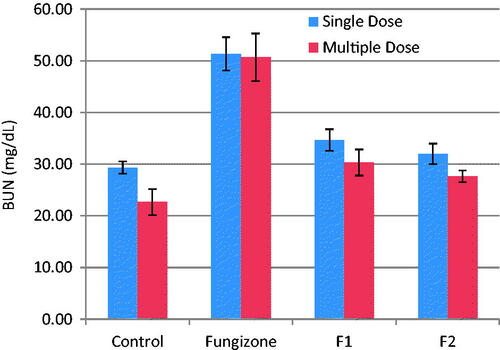
Figure 4. Plasma Cr, mg/dL, (mean ± SD) after single and multiple iv doses (5 mg/kg) of AMB and AMB loaded to PLGA-PEG NP to rats (n = 3).
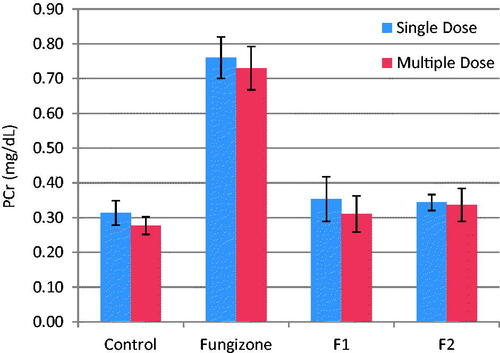
PCr were in the range of 0.31–0.34 mg/dl for both formulations, which is within the normal range for rat plasma. Therefore, AMB loaded to PLGA-PEG NPs, showed minimal renal effects than Fungizone® over the short duration of treatment.
The histopathological analysis of kidney tissue after iv administrations of the NPs formulations of AMB and Fungizone®, did not confirm any unusual sign of necrosis in all treated groups except in the Fungizone® treated group. It showed a distinctive necrosis of various degree as presented in , which has been approved by the PCr results in the same study. As a conclusion, all the iv administrations of the developed AMB-loaded to PLGA–PEG Diblock copolymer investigated, showed minimal renal damage compared to the reference formulation Fungizone®, over a short duration of treatment.
Figure 5. Typical kidney tissue alterations verified in rats treated with AMB or its equivalent dose as 1.0 mg/kg of body weight as iv administration of different Amb-PLGAPEG copolymer. 1) normal kidney tissue; 2,3,4) Fungizone® f1 and f2, respectively, varying degree of nephrotoxicity necrosis related to iv administration of iv doses (5 mg/kg).
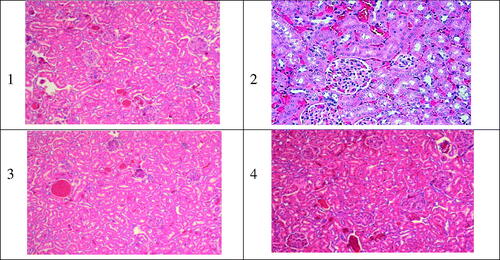
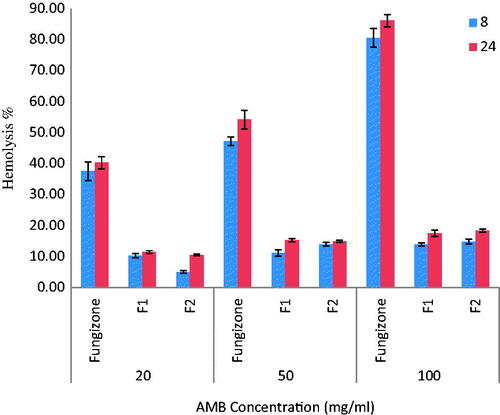
Figure 6. In vitro mean RBCs hemolysis following incubation of RBC with Fungizone® and different AMB-NPs formulations at concentrations of 20, 50 and 100 μg/ml (n = 3).
In vitro hemolysis test
In order to determine the effect of the NPs of AMB on hemolytic profile, the RBS lysis induced by AMB NP selected formulations were compared with the Fungizone®.
The extent of hemolysis induced after incubation of RBCs with F1 and F2 formulations of AMB-NP in comparison with Fungizone® is depicted in . The hemolysis was high (84%) in the case of Fungizone® at 100 μg/ml and 43% at 20 μg/ml. Therefore, it seems that Fungizone® is likely to be toxic, even at the lowest concentration used in the experiment. At a similar concentration AMB loaded to PLGA–PEG NPs formulations showed negligible hemolysis. The results in also indicate that the hemolysis was dose-dependent. Regarding the degree of hemolysis, the tested formulations were classified as low hemolytic toxicity 4 to 8 times hemolysis reduction in comparison with Fungizone®, F2 formulation showed the lowest hemolysis reflects a better control over the rate of AMB diffusion from these selective formulations over the Fungizone®. After 24 h incubation, similar results were obtained as that after 8 h incubation, indicating that the releases of AMB from these formulations is slow and it seems that PLGA-PEG NP imparts a protective effect to AMB in preventing the RBC lysis of these formulations.
In vitro antifungal activity
shows the antimicrobial activity of pure AMB, Fungizone® and the selected formulations of AMB loaded to PLGA-PEG NP in C. albicans after 24 and 48 h incubations. The MIC-0 for pure AMB was found to be 0.5 μg/ml after 24 h and 1.0 μg/ml after 48 h incubation. Similar results are obtained by others (Nishi et al., Citation2007). Meanwhile, the MIC-0 for AMB loaded to PLGA–PEG NP formulations was reduced by ≥ fourfold.
Table 5. Checkboard assay of AMB against C. albicans (n = 6).
Discussion
Several reports show that parenteral administrations of AMB polymeric nanoparticles formulations, have antifungal efficacy and lower toxicity with increase accessibility of the drug to organs and targeted tissue (Amaral et al., Citation2009; Laniado-Laborín & Cabrales-Vargas, Citation2009; Souza et al., Citation2015). It was also reported that some carriers added during the formulation process affect the aggregation state and hence AMB activity (Legrand et al., Citation1992). Barwicz et al. have demonstrated the ability of surfactants to reduce the toxicity of AMB via decreasing the aggregation state of the drug (Barwicz et al., Citation1992; Adams and Kwon, Citation2003).
Currently, PLAG and PLGA-PEG are being used to improve the release rate of different classes of drugs from NPs systems intended for both parenteral (Allémann et al., Citation1998; Huh et al., Citation2003; Packhaeuser et al., Citation2004; Cheng et al., Citation2007; Saadati & Dadashzadeh, Citation2014; Teekamp et al., Citation2015) and oral administrations (Yoo & Park, Citation2001; Garinot et al., Citation2007; Fernandez-Carballido et al., Citation2008; Khalil et al., Citation2013; Yan et al., Citation2015). The ability of PLGA–PEG to enhance bioavailability of hydrophobic drugs, has been demonstrated for different classes of drugs. Curcumin potential efficacy is limited by its lack of solubility in aqueous solvents and poor oral bioavailability. PLGA–PEG NPs were able to increase curcumin bioavailability by 3.5-fold compared to curcumin loaded PLGA NPs alone (Anand et al., Citation2010).
GA and its derivatives have been utilized in enhancing drug absorption (Tanaka et al., Citation1992; Imai et al., Citation1999; Radwan & Aboul-Enein, Citation2002; Cho et al., Citation2004; Anand et al., Citation2010). Significant enhanced rectal absorption of AMB suppositories in rabbits following GA addition in comparison to formulation containing no GA (Tanaka et al., Citation1992; Anand et al., Citation2010). Insulin nasal spray formulations for nasal delivery containing GA showed improved bioavailability (Khalil et al., Citation2013). Additionally oral delivery of heparin is achieved by addition of GA (Motlekar et al., Citation2006).
Comparing the relative bioavailability of each tested formulation to F1-PO without GA, resulted in a maximum improvement of 798% in Frel to Fungizone, which is better than the outcome of the published investigations as presented in . Sachs-Barrable et al. (Citation2008) was exceptional in improving the relative F of AMB but in different system and dose of Fungizone® used.
Table 6. Relative bioavailability of AMB in rats, after PO administration of various AMB-loaded NPs in different formulations, in comparison to Fungizone®.
AMB nephrotoxicity is manifested by renal insufficiency due to glomerular and vascular disease in addition to abnormalities in tubular function. Acute AMB-induced nephrotoxicity is characterized by increased BUN and increased PCr levels. BUN is an important biochemical marker, which is routinely evaluated as an indicator of clinical renal function. Abnormally high BUN levels indicate the dysfunction or damage to the kidney. Similarly, PCr is another reliable marker of renal function. Creatinine, the end product of muscle metabolism, is filtered and excreted by the kidneys. Elevated PCr levels is indicative of renal dysfunction (Italia et al., Citation2009; Tonomura et al., Citation2009).
The reduced toxicity of AMB-NP observed could be due to the fact that AMB was slowly released from AMB-NP than from the deoxycholate micelles of Fungizone®. AMB is known to induce vasoconstriction, which leads to a decrease of blood flow in the kidney. The observed reduction in the PCr is believed to be caused by a decrease in blood pressure that is associated with a decrease in blood flow in the glomerulus (Tasset et al., Citation1992; Tiyaboonchai et al., Citation2001; Risovic et al., Citation2003; Nahar et al., Citation2008; Italia et al., Citation2009; Tonomura et al., Citation2009; Belkherroubi-Sari et al., Citation2011).
It is reported that AMB is highly toxic in its aggregated state than in its monomer form (Brajtburg & Bolard, Citation1996; Nishi et al., Citation2007). In solution, AMB exists in three different forms; monomers, oligomers and aggregates. The soluble form of AMB exists in monomeric form (Brajtburg & Bolard, Citation1996; Nishi et al., Citation2007).
The ratio of absorbance at 348 nm to 409 nm is reported to give the extent of aggregation in AMB (Legrand et al., Citation1992). Barwicz et al. (Citation1992) report the ratio to be 2 for aggregated species. Most of the AMB formulations have greater A348/A409 values. For instance, Fungizone® has a value of 2.9 while Ambisome® has a value of 4.8 (Mullen et al., Citation1997). For AMB-loaded PLGA-PEG, the value obtained was less than1.14 showing that AMB was not aggregated in the novel oral formulation. Therefore, assessment of hemolytic toxicity seems to be a prerequisite while developing any formulation of AMB.
The lack of hemolysis activity may reflect the release of monomeric AMB from AMB-loaded PLGA-PEG copolymer as opposed to Fungizone®, which release both aggregated and monomeric forms of the drug (Yu et al., Citation1998; Adams & Kwon, Citation2003; Brajtburg & Bolard, Citation1996). Therefore, the low hemolysis of all AMB-NP as shown in might be attributed to the encapsulation of a nanoaggregate form of AMB, and slow diffusion of AMB from the conjugated polymer. Another explanation, is that during formulation, AMB was added in an aqueous polymer phase that was acidified with 5N HCL (pH 3) (far from the pKa of the drug; 3.7 and 10), thus precluding self-association or aggregation.
Fungizone® caused more extent of hemolysis because Fungizone® formulation consist of micellar dispersion of AMB with sodium deoxycholate, which act as surfactant and can induce hemolysis itself in addition to the hemolysis caused by AMB. Fungizone® is well-known for causing hemolysis mainly due to active hemolysis by pore formation and changing electrolyte balance in erythrocytes (Yu et al., Citation1998; Fukui et al., Citation2003; Bang et al., Citation2008; Nahar et al., Citation2008; Italia et al., Citation2009; Falamarzian & Lavasanifar, Citation2010; Jain & Kumar, Citation2010; Shao et al., Citation2010; Sheikh et al., Citation2010; Asghari, Citation2011).
AMB at concentrations > 0.5 to 1-fold the MIC has a fungicidal activity, while AMB at concentrations < 0.5 to 1-fold the MIC has a fungistatic activity, a finding that has been described previously (Tasset et al., Citation1992; Pfaller & Barry, Citation1994; Risovic et al., Citation2003; Belkherroubi-Sari et al., Citation2011). The MIC of AMB loaded to PLGA-PEG NP against C. albicans was reduced two to three-fold compared with free AMB. Therefore, the prepared AMB-NPs formulations have high therapeutic efficacy and are useful for the treatment of fungal infection including candidiasis.
Conclusion
In conclusion, an innovative AMB oral formulation was developed which would improve the patient adherence due to the potential reduction of AMB adverse side effects especially nephrotoxicity and induction of hemolysis. PEGylated polymer (PLGA-PEG) enhances the oral absorption of AMB compared to Fungizone®. On a cellular level, AMB loaded to PLGA-PEG copolymer showed potent activity against in vitro C. albicans with no pathological abnormalities were observed in rats’ kidney tissues. A further study is needed to investigate the in vivo efficacy of the developed formulation.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
This research project was supported by a grant from the “Research Center of the Female Scientific and Medical Colleges”, Deanship of Scientific Research, King Saud University.
Related Research Data
References
- Adams ML, Kwon GS. (2003). Relative aggregation state and hemolytic activity of amphotericin B encapsulated by poly (ethylene oxide)-block–poly (N-hexyl-l-aspartamide)-acyl conjugate micelles: effects of acyl chain length. J Control Release 87:23–32
- Al-Quadeib BT, Radwan MA, Siller L, et al. (2015). Stealth amphotericin B nanoparticles for oral drug delivery: in vitro optimization. Saudi Pharm J 23:290–302
- Al‐Quadeib BT, Radwan MA, Siller L, et al. (2014). Therapeutic monitoring of amphotericin B in Saudi ICU patients using UPLC MS/MS assay. Biomed Chromat. 28:1652–9
- Allémann E, Leroux J, Gurny R. (1998). Biodegradable nanoparticles of poly (lactic acid) and poly (lactic-co-glycolic acid) for parenteral administration. Pharm Dosage Forms: Disperse Syst 3:163–93
- Amaral AC, Bocca AL, Ribeiro AM, et al. (2009). Amphotericin B in poly (lactic-co-glycolic acid)(PLGA) and dimercaptosuccinic acid (DMSA) nanoparticles against paracoccidioidomycosis. J Antimicrob Chemother 63:526–33
- Anand P, Nair HB, Sung B, et al. (2010). Design of curcumin-loaded PLGA nanoparticles formulation with enhanced cellular uptake, and increased bioactivity in vitro and superior bioavailability in vivo. Biochem Pharmacol 79:330–8
- Asghari H. (2011). Preparation and antifungal activity of spray-dried amphotericin B-loaded nanospheres. DARU J Pharm Sci 19:351–5
- Bang JY, Song CE, Kim C, et al. (2008). Cytotoxicity of amphotericin B-incorporated polymeric micelles composed of poly (DL-lactide-co-glycolide)/dextran graft copolymer. Arch Pharm Res 31:1463–9
- Barwicz J, Christian S, Gruda I. (1992). Effects of the aggregation state of amphotericin B on its toxicity to mice. Antimicrob Agents Chemother 36:2310–15
- Bekersky II, Fielding RM, Buell D, Lawrence II. (1999). Lipid-based amphotericin B formulations: from animals to man. Pharm Sci Technol Today 2:230–6
- Belkherroubi-Sari L, Boucherit Z, Boucherit K, Belbraouet S. (2011). Study of renal toxicity in wistar rats following the action of amphotericin B solution prepared under extreme pH conditions. Food Nutri Sci 2:731–5
- Brajtburg J, Bolard J. (1996). Carrier effects on biological activity of amphotericin B. Clin Microbiol Rev 9:512–31
- Chen LY, Davey AK, Chen YX, et al. (2009). Effects of diammonium glycyrrhizinate on the pharmacokinetics of aconitine in rats and the potential mechanism. Xenobiotica 39:955–63
- Cheng J, Teply BA, Sherifi I, et al. (2007). Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 28:869–76
- Cho SW, Lee JS, Choi SH. (2004). Enhanced oral bioavailability of poorly absorbed drugs. I. Screening of absorption carrier for the ceftriaxone complex. J Pharm Sci 93:612–20
- Chuealee R, Aramwit P, Noipha K, Srichana T. (2011). Bioactivity and toxicity studies of amphotericin B incorporated in liquid crystals. Eur J Pharm Sci 43:308–17
- Cifani C, Costantino S, Massi M. (2012). Commercially available lipid formulations of amphotericin B: are they bioequivalent and therapeutically equivalent? Acta Bio Medica Atenei Parmensis 83:154–63
- Deray G. (2002). Amphotericin B nephrotoxicity. J Antimicrob Chemother 49:37–41
- Drew RH. (2013). Pharmacology of amphotericin B [Online]. Wolter Kluwer. Available at: www.uptodate.com/contents/pharmacology-of-amphotericin-b [last accessed 26 Apr 2013]
- Egger P, Bellmann R, Wiedermann CJ. (2001). Determination of amphotericin B, liposomal amphotericin B, and amphotericin B colloidal dispersion in plasma by high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl 760:307–13
- Falamarzian A, Lavasanifar A. (2010). Chemical modification of hydrophobic block in poly(ethylene oxide) poly(caprolactone) based nanocarriers: effect on the solubilization and hemolytic activity of amphotericin B. Macromol Biosci 10:648–56
- Fernandez-Carballido A, Pastoriza P, Barcia E, et al. (2008). PLGA/PEG-derivative polymeric matrix for drug delivery system applications: characterization and cell viability studies. Int J Pharm 352:50–7
- Fukui H, Koike T, Saheki A, et al. (2003). Evaluation of the efficacy and toxicity of amphotericin B incorporated in lipid nano-sphere (LNS®). Int J Pharm 263:51–60
- Garinot M, Fiévez V, Pourcelle V, et al. (2007). PEGylated PLGA-based nanoparticles targeting M cells for oral vaccination. J Control Release 120:195–204
- Gershkovich P, Sivak O, Wasan EK, et al. (2010). Biodistribution and tissue toxicity of amphotericin B in mice following multiple dose administration of a novel oral lipid-based formulation (iCo-009). J Antimicrob Chemother 65:2610–13
- Gibaldi & Perrier. 1982. Pharmacokinetics, New York: Marcel Dekker
- Huh KM, Cho YW, Park K. (2003). PLGA-PEG block copolymers for drug formulations. Drug Deliv Technol 3:42–4
- Ibrahim F, Gershkovich P, Sivak O, et al. (2012). Assessment of novel oral lipid-based formulations of amphotericin B using an in vitro lipolysis model. Eur J Pharm Sci 46:323–8
- Imai T, Sakai M, Ohtake H, et al. (1999). In vitro and in vivo evaluation of the enhancing activity of glycyrrhizin on the intestinal absorption of drugs. Pharm Res 16:80–6
- Italia J, Yahya M, Singh D, Kumar MR. (2009). Biodegradable nanoparticles improve oral bioavailability of amphotericin B and show reduced nephrotoxicity compared to intravenous Fungizone®. Pharm Res 26:1324–31
- Italia JL, Sharp A, Carter KC, et al. (2011). Peroral amphotericin B polymer nanoparticles lead to comparable or superior in vivo antifungal activity to that of intravenous Ambisome® or Fungizone™. PloS One 6:1–8
- Jain JP, Kumar N. (2010). Development of amphotericin B loaded polymersomes based on (PEG) 3-PLA co-polymers: factors affecting size and in vitro evaluation. Eur J Pharm Sci 40:456–65
- Jain S, Valvi PU, Swarnakar NK, Thanki K. (2012). Gelatin coated hybrid lipid nanoparticles for oral delivery of amphotericin B. Mol Pharm 9:2542–53
- Kayser O, Olbrich C, Yardley V, et al. (2003). Formulation of amphotericin B as nanosuspension for oral administration. Int J Pharm 254:73–5
- Khalil NM, Do Nascimento TCF, Casa DM, et al. (2013). Pharmacokinetics of curcumin-loaded PLGA and PLGA-PEG blend nanoparticles after oral administration in rats. Colloids Surf B Biointerfaces 101:353–60
- Kumar R, Sahoo GC, Pandey K, et al. (2015). Study the effects of PLGA-PEG encapsulated Amphotericin B nanoparticle drug delivery system against Leishmania donovani. Drug Deliv 22:383–8
- Laniado-Laborín R, Cabrales-Vargas MN. (2009). Amphotericin B: side effects and toxicity. Revista Iberoamericana De Micología 26:223–7
- Legrand P, Romero EA, Cohen BE, Bolard J. (1992). Effects of aggregation and solvent on the toxicity of amphotericin B to human erythrocytes. Antimicrob Agents Chemother 36:2518–22
- Miller C, Waller E, Klingemann H, et al. (2004). Lipid formulations of amphotericin B preserve and stabilize renal function in HSCT recipients. Bone Marrow Transplant 33:543–8
- Motlekar NA, Srivenugopal KS, Wachtel MS, Youan BBC. (2006). Evaluation of the oral bioavailability of low molecular weight heparin formulated with glycyrrhetinic acid as permeation enhancer. Drug Develop Res 67:166–74
- Mullen A, Carter K, Baillie A. (1997). Comparison of the efficacies of various formulations of amphotericin B against murine visceral leishmaniasis. Antimicrob Agents Chemother 41:2089–92
- Nahar M, Jain NK. (2009). Preparation, characterization and evaluation of targeting potential of amphotericin B-loaded engineered PLGA nanoparticles. Pharm Res 26:2588–98
- Nahar M, Mishra D, Dubey V, Jain NK. (2008). Development, characterization, and toxicity evaluation of amphotericin B–loaded gelatin nanoparticles. Nanomed Nanotechnol Biol Med 4:252–61
- Nishi K, Antony M, Mohanan P, et al. (2007). Amphotericin B-gum arabic conjugates: synthesis, toxicity, bioavailability, and activities against Leishmania and fungi. Pharm Res 24:971–80
- Packhaeuser C, Schnieders J, Oster C, Kissel T. (2004). In situ forming parenteral drug delivery systems: an overview. Eur J Pharm Biopharm 58:445–55
- Pfaller M, Barry A. (1994). Evaluation of a novel colorimetric broth microdilution method for antifungal susceptibility testing of yeast isolates. J Clin Microbiol 32:1992–1996
- Pompei R, Pani A, Flore O, et al. (1980). Antiviral activity of glycyrrhizic acid. Experientia 36:304
- Prajapati VK, Awasthi K, Yadav TP, et al. (2011). An oral formulation of amphotericin B attached to functionalized carbon nanotubes is an effective treatment for experimental visceral leishmaniasis. J Infect Dis 205:333–6
- Radwan MA, Aboul-Enein HY. (2002). The effect of oral absorption enhancers on the in vivo performance of insulin-loaded poly (ethylcyanoacrylate) nanospheres in diabetic rats. J Microencapsul 19:225–235
- Risovic V, Boyd M, Choo E, Wasan KM. (2003). Effects of lipid-based oral formulations on plasma and tissue amphotericin B concentrations and renal toxicity in male rats. Antimicrob Agents Chemother 47:3339–3342
- Saadati R, Dadashzadeh S. (2014). Marked effects of combined TPGS and PVA emulsifiers in the fabrication of etoposide-loaded PLGA-PEG nanoparticles: in vitro and in vivo evaluation. Int J Pharm 464:135–144
- Sabra R, Branch RA. (1990). Amphotericin B nephrotoxicity. Drug Safety 5:94–108
- Sachs-Barrable K, Lee SD, Wasan EK, et al. (2008). Enhancing drug absorption using lipids: a case study presenting the development and pharmacological evaluation of a novel lipid-based oral amphotericin B formulation for the treatment of systemic fungal infections. Adv Drug Deliv Rev 60:692–701
- Serrano DR, Lalatsa A, Dea-Ayuela MA, et al. (2015). Oral particle uptake and organ targeting drives the activity of amphotericin B nanoparticles. Mol Pharm 12:420–431
- Shao K, Huang R, Li J, et al. (2010). Angiopep-2 modified PE-PEG based polymeric micelles for amphotericin B delivery targeted to the brain. J Control Release 147:118–126
- Sheikh S, Ali SM, Ahmad MU, et al. (2010). Nanosomal amphotericin B is an efficacious alternative to Ambisome® for fungal therapy. Int J Pharm 397:103–108
- Singh K, Tiwary A, Rana V. (2013). Spray dried chitosan–EDTA superior microparticles as solid substrate for the oral delivery of amphotericin B. Int J Biol Macromol 58:310–319
- Sivak O, Gershkovich P, Lin M, et al. (2011). Tropically stable novel oral lipid formulation of amphotericin B (iCo-010): biodistribution and toxicity in a mouse model. Lipids Health Dis 10:135
- Skiba-Lahiani M, Hallouard F, Mehenni L, et al. (2015). Development and characterization of oral liposomes of vegetal ceramide based amphotericin B having enhanced dry solubility and solubility. Mater Sci Eng C 48:145–149
- Souza A, Nascimento A, De Vasconcelos N, et al. (2015). Activity and in vivo tracking of amphotericin B loaded PLGA nanoparticles. Eur J Med Chem 95:267–276
- Standards (NCFCL). (2002). Reference methods for broth dilution antifungal susceptibility testing of yeast: Approved standard, National Committee for Clinical Laboratory Standards
- Sundar S, Chakravarty J, Agarwal D, et al. (2010). Single-dose liposomal amphotericin B for visceral leishmaniasis in India. N Engl J Med 362:504–512
- Tanaka M, Takahashi M, Kuwahara E, et al. (1992). Effect of glycyrrhizinate on dissolution behavior and rectal absorption of amphotericin B in rabbits. Chem Pharm Bull 40:1559–1562
- Tasset C, Preat V, Bernard A, Roland M. (1992). Comparison of nephrotoxicities of different polyoxyethyleneglycol formulations of amphotericin B in rats. Antimicrob Agents Chemother 36:1525–1531
- Teekamp N, Duque LF, Frijlink HW, et al. (2015). Production methods and stabilization strategies for polymer-based nanoparticles and microparticles for parenteral delivery of peptides and proteins. Expert Opin Drug Deliv 12:1311–31
- Tiyaboonchai W, Woiszwillo J, Middaugh CR. (2001). Formulation and characterization of amphotericin B-polyethylenimine-dextran sulfate nanoparticles. J Pharm Sci 90:902–914
- Tonomura Y, Yamamoto E, Kondo C, et al. (2009). Amphotericin B-induced nephrotoxicity: characterization of blood and urinary biochemistry and renal morphology in mice. Hum Exp Toxicol 28:293–300
- Torrado J, Espada R, Ballesteros M, Torrado‐Santiago S. (2008). Amphotericin B formulations and drug targeting. J Pharm Sci 97:2405–2425
- Tozer TN, Rowland M. (2006). Introduction to pharmacokinetics and pharmacodynamics: the quantitative basis of drug therapy. Philadelphia, PA: Lippincott Williams & Wilkins
- Yan Q, Xiao LQ, Tan L, et al. (2015). Controlled release of simvastatin‐loaded thermo‐sensitive PLGA‐PEG‐PLGA hydrogel for bone tissue regeneration: in vitro and in vivo characteristics. J Biomed Mater Res Part A 103:3508–9
- Yang FH, Zhang Q, Liang QY, et al. (2015). Bioavailability enhancement of paclitaxel via a novel oral drug delivery system: paclitaxel-loaded glycyrrhizic acid micelles. Molecules 20:4337–4356
- Yang Z, Chen M, Yang M, et al. (2014). Evaluating the potential of cubosomal nanoparticles for oral delivery of amphotericin B in treating fungal infection. Int J Nanomed 9:327
- Yang Z, Tan Y, Chen M, et al. (2012). Development of amphotericin B-loaded cubosomes through the solemuls technology for enhancing the oral bioavailability. AAPS PharmSciTech 13:1483–1491
- Yoo HS, Park TG. (2001). Biodegradable polymeric micelles composed of doxorubicin conjugated PLGA-PEG block copolymer. J Control Release 70:63–70
- Yu B, Okano T, Kataoka K, Kwon G. (1998). Polymeric micelles for drug delivery: solubilization and haemolytic activity of amphotericin B. J Control Release 53:131–136
- Zu Y, Sun W, Zhao X, et al. (2014). Preparation and characterization of amorphous amphotericin B nanoparticles for oral administration through liquid antisolvent precipitation. Eur J Pharm Sci 53:109–117