
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Oligopeptide transporter 1 (PepT1) has been a striking prodrug-designing target. However, the underlying mechanism of PepT1 as a target to facilitate the oral absorption of nanoparticles (NPs) remains unclear. Herein, we modify Poly (lactic-co-glycolic acid) (PLGA) NPs with the conjugates of dipeptides (L-valine-valine, L-valine-phenylalanine) and polyoxyethylene (PEG Mw: 1000, 2000) stearate to facilitate oral delivery of docetaxel (DTX) to investigate the oral absorption mechanism and regulatory effects on PepT1 of the dipeptide-modified NPs. The cellular uptake of the dipeptide-modified NPs is more efficient than that of the unmodified NPs in the stably transfected hPepT1- Hela cells and Caco-2 cells, suggesting the involvement of PepT1 in the endocytosis of NPs. The internalization of the dipeptide-modified NPs is proved to be a proton-dependent process. Moreover, the L-valine-valine modified NPs with shorter PEG chain exhibit distinct advantages in terms of intestinal permeability and oral absorption, resulting in significantly improved oral bioavailability of DTX. In summary, PepT1 could serve as a desirable target for oral nanoparticulate drug delivery and the dipeptide-modified NPs represent a promising nanoplatform to facilitate oral delivery of hydrophobic drugs with low bioavailability.
Introduction
Oral administration has been considered as a preferred route in clinical treatment due to its convenience, patient compliance, and lower-cost. Over the decades, enormous developments in nanotechnology have introduced new approaches to improve oral drug delivery (Shan et al., Citation2016; Fan et al., Citation2018; Qu et al., Citation2018). However, the ideal oral delivery efficacy still confronts great challenges, including the poor physicochemical properties of drugs, harsh gastrointestinal environment, the diffusion barrier of intestinal mucus layer, and the compromised enterocyte absorption before entering systemic circulation (Shan et al., Citation2015; Tang et al., Citation2018).
Effort has been made to address these challenges. Among all the strategies, targeting delivery, especially membrane transporters are increasingly becoming striking targets for drug delivery (Li et al., Citation2013; Jiang et al., Citation2014; Tao et al., Citation2015; An et al., Citation2016; Tao et al., Citation2016; Zhu et al., Citation2016; Rosenblum et al., Citation2018). Transporters are integral transmembrane proteins and play a key role in the transport of ions, small molecules, and macromolecules across biomembrane (Luo et al., Citation2016; Kou et al., Citation2018). In general, transporters could be mainly categorized into two major super families including solute carriers (SLC) and ATP-binding cassette (ABC) depending on whether there is direct ATP consumption. Moreover, they usually prefer low molecule-weight substrates or analogs, which are easy to modify, more stable in gastrointestinal tract, less immunogenic, and more flexible when reaching the binding site due to less steric hindrance (Pereira et al., Citation2014).
The widely distributed intestinal transporters provide new possibilities for oral drug delivery (Kou et al., Citation2017). Among them, oligopeptide transporter 1 (PepT1) has been an attracting prodrug design target for delivering hydrophilic drugs with poor membrane permeability (Zhang et al., Citation2013b). PepT1 belongs to the SLCs family, which is also known as SLC15A1. It is a transport protein with high transport capacity and low substrate affinity and has the highest level of protein abundance compared with other transporters expressed along the small intestine (Drozdzik et al., Citation2014). Physiologically, it could transport di/tri peptides, angiotensin-converting enzyme inhibitors (ACEi), β-lactam antibiotics and other peptidomimetic agents from intestinal lumen to enterocytes driven by an inward proton-electrochemical gradient (Han et al., Citation2015). Our group has previously reported several PepT1 targeting prodrugs (Sun et al., Citation2009; Yan et al., Citation2011; Zhang et al., Citation2013a), inspired by the successful exploitation of valine modified prodrug valacyclovir (Ganapathy et al., Citation1998) and valganciclovir (Sugawara et al., Citation2000). In all cases, we found that the valine modified peptidomimetics exhibited best improvement in bioavailability, while the phenylalanine modified prodrug demonstrated favorable absorption as well. It was also reported the valine-valine modified NO-donating oleanolic acid derivative had higher oral bioavailability than other dipeptide modified prodrugs (Fang et al., Citation2014). Besides, L-type di/tripeptides and peptidomimetics were found to have preferred membrane permeability and higher binding affinity compared with D-type substrates. Although PepT1 has been widely used as a target for small molecule prodrugs, the role of PepT1 as a target to facilitate nanoparticle (NPs) internalization and its underlying mechanism hasn’t been investigated until now.
Herein, we coupled dipeptide (L-valine-valine, L-valine-phenylalanine) to the amphiphilic polyoxyethylene (PEG) stearate to synthesize PepT1-targeted polymers. Different molecular weight of polyoxyethylene stearate (PEG Mw: 1000, 2000) were introduced into PepT1-targeted polymers for targeting modification on PLGA NPs to investigate the influence of outer hydrophilic spacer on PepT1 mediated endocytosis (Tao et al., Citation2013, Citation2017a), taking advantage of its ability to improve the oral absorption of NPs (Ramanathan et al., Citation2001). The modified NPs were loaded with docetaxel (DTX), whose oral delivery efficacy was severely limited by low solubility and permeability. We found that the L-valine-valine and polyoxyethylene stearate (PEG Mw: 1000) polymer modified NPs displayed higher PepT1 targeting efficiency than others. Our results demonstrated that the dipeptide modification could facilitate the PepT1-mediated cellular uptake of the modified NPs in a proton-dependent manner, resulting in significantly improved oral absorption of DTX. Furthermore, the underlying regulatory mechanism of the modified NPs on PepT1 expression and distribution was also elucidated, laying a good foundation for the further investigation and clinical application of PepT1-based nanoparticulate drug delivery system.
Materials and methods
Materials
Polyoxyethylene stearate 25 (SP25) were purchased from Aladdin (Shanghai, China). Polyoxyethylene stearate 40 (SP40) and coumarin-6 (C6) were obtained from Sigma (St. Louis, MO). N-carbobenzyloxy-L-valine-valine (Cbz-L-val-val) and N-carbobenzyloxy-L-valine-phenylalanine (Cbz-L-val-phe) were gained from GL Biochem Ltd. (Shanghai China). Poly (lactic-co-glycolic acid) (PLGA, lactide:glycolide =75:25, Mw 38,000) was purchased from Daigang Biomaterial Co., Ltd. (Jinan, China). Polyvinyl alcohol (PVA, Mw 20,000–30,000) was gained from Across Organics, Thermo Fisher Scientific, NJ. Docetaxel (DTX) was obtained from Nanjing Jingzhu Bio-Technology Co. Ltd. (Nanjing, China). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) was purchased from Shanghai Haiqu Pharm Co., Ltd. 4-Dimethylaminopyridine (DMAP) was gained from Shanghai Medpep Co., Ltd. (Shanghai, China). Glycylsarcosine (GlySar) and trypsin-EDTA was purchased from Melone Pharmaceutical Co., Ltd. (Dalian, China). Fetal bovine serum (FBS) and Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA)) were purchased from Gibco BRL (Gaithersburg, MD).
Synthesis of dipeptide-modified PepT1-targeting polymer
Dipeptides (L-valine-valine, L-valine-phenylalanine) and polyoxyethylene stearate conjugates were synthesized by a two-step reaction including esterification and hydrogenation reduction. Cbz-L-val-val and Cbz-L-val-phe were coupled to polyoxyethylenen stearate (n = 25, 40, representing PEG Mw: 1000, 2000) in the presence of catalysts EDC and DMAP. After column purification, Cbz group was removed by Pd/C. The chemical structures of the products were confirmed by proton nuclear magnetic resonance (1H-NMR). The resultant polymers L-val-val-polyoxyethylene stearate1000, L-val-val-polyoxyethylene stearate2000, L-val-phe-polyoxyethylene stearate1000, and L-val-phe-polyoxyethylene stearate2000 were denoted as NSPV1000, NSPV2000, NSPP1000, and NSPP2000 in the following experiments.
Preparation and characterization of dipeptide-modified NPs
DTX-loaded modified NPs were prepared by emulsion-solvent evaporation method. Briefly, a mixture of PLGA, DTX (or C6), and dipeptide-polyoxyethylene stearate conjugates (20:1:2, weight ratio) were dissolved in 1 mL dichloromethane and was subsequently added into 5 mL 1% PVA solution. The two-phase mixture was emulsified by sonicating for 5 min at 400 W in an ice water bath and then evaporated for 5 h under room temperature. The final NPs suspensions were centrifuged to remove the unencapsulated DTX. Similarly, the unmodified NPs were prepared using the same method mentioned above, except the dipeptide-polyoxyethylene stearate conjugates were replaced by polyoxyethylene stearate. The drug loading capacity (DL%) and encapsulation efficiency (EE%) were measured using High-performance liquid chromatography (HPLC) at 227 nm with a method described previously (Li et al., Citation2016).
The surface nitrogen contents of NSPV1000 NPs (with a series dipeptide densities of 5, 10, and 20%, weight ratio) were measured by X-ray photoelectron spectroscopy (ESCALAB250, Thermo VG, Waltham, MA.), using the unmodified NPs as a control. The particle size and zeta potential were measured by dynamic light scattering (DLS; Nano ZS, Malvern, Co., U.K.). All measurements were performed triplicates. The morphology of NSPV1000 NPs was also negatively stained and visualized using Transmission Electron Microscope (TEM; Tecnai G20, FEI, Hillsboro, OR). In addition, X-ray diffraction (D/Max 2500 PC, Rigaku, Japan) was used to assess the molecule status of NSPV1000 NPs, blank NPs, physical mixtures, and free DTX. The colloidal stability study of NSPV1000 NPs was conducted in the presence of pH 1.0, 6.8, and 7.4 solution, simulated gastric fluid, simulated intestinal fluid, and 10% FBS. The variation of particle size was monitored at predetermined time points.
The in vitro drug release of DTX from DTX solution, unmodified NPs, NSPV1000 NPs, NSPV2000 NPs, NSPP1000 NPs, and NSPP2000 NPs were conducted by dialysis method. Briefly, 200 µL of each DTX formulations was placed in pretreated dialysis bag (MWCO: 8–10 kDa) which was immersed in 30 mL phosphate buffered saline (PBS; pH 1.0 and 6.8) containing 0.5% Tween-80 (w/v). The samples were incubated under 37 °C with a shaking speed of 100 rpm. Two milliliter aliquots of the release medium were withdrawn and replaced with an equivalent volume of fresh medium. The DTX concentration of the collected samples were analyzed using the same method described above.
Cell culture
Hela cells stably transfected with human PepT1 plasmid (hPepT1-Hela) and vector (mock-Hela) cells were kindly provided by Professor Kexin Liu (Dalian Medical University, Dalian, China). The cells were maintained in high-glucose DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin (Guo et al., Citation2012, Citation2014). Porcine ileum Caco-2 cells were obtained from American Type Culture Collection (Rockvile, MD) and cultured in Modified Eagle’s medium (MEM; Invitrogen, Carlsbad, CA) containing 10% FBS, 1 mM sodium pyruvate, 1% non-essential amino acids, 1% L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. All the cells were grown at 37˚C with an atmosphere of 5% CO2 and 95% relative humidity.
Western blotting of PepT1 expression in cells
The hPepT1-Hela and mock-Hela cells were collected and lysed by radio immuno precipitation assay buffer (RIPA) including 1 mM phenylmethylsulfonyl fluoride (PMSF). The protein concentration was measured by BCA (bicinchoninic acid) assay kit (Dingguo Changsheng Biotech, Beijing) and 30 μg of protein extracts were loaded on 10% SDS-PAGE gel for electrophoresis. The separated gel was then transferred and blotted to 0.22 um polyvinylidene fluoride membranes. Then the membranes were blocked with 5% bovine serum albumin (BSA) in TBST (10 mM Tris-HCl, 150 mM NaCl and 0.1% Tween-20) for 1 h and subsequently incubated with primary antibody for PepT1 (sc-20653, Santa Cruz, Dallas, TX) overnight at 4 °C. β-Actin was blotted as control. The positive protein bands were visualized by ChemiDocTM XRS+Imaging system (Bio-Rad, Hercules, CA). Protein expression was quantified by Image LabTM Software (Bio-Rad).
Cellular uptake
C6 was incorporated in the NPs to evaluate the cellular uptake efficacy. The hPepT1-Hela and mock-Hela cells were seeded in 24-well plates (Corning, Tokyo, Japan) at a density of 5 × 104 cells/well. After 24 h of attachment, the cells were treated with 1 mL fresh Hank’s buffered salt solution (HBSS) containing various C6 formulations (C6 solution, unmodified and dipeptide modified NPs, with same C6 concentration of 5 μg/mL), with or without the presence of 20 mM typical substrate GlySar for 1 and 3 h at 37 °C. The uptake study was also conducted under modified conditions (with the cultured medium adjusted to pH 5.5, 6.5, and 7.4). Cell morphology under different pH conditions was monitored by microscope after the experiments. After incubation, the cell layer was washed with cold PBS for three times and then was lysed in 100 μL RIPA buffer for 1 h in the shaker. The fluorescence intensity of internalized C6 was analyzed by microplate reader with an excitation/emission wavelength set at 458 nm/525 nm and the protein concentration was measured by BCA assay kit. Uptake profile of C6 formulations in Caco-2 cells were similarly examined, except the cells were allowed to grow for 14 days before the study, with the medium changed every other day.
Regulatory mechanism of the modified NPs on PepT1 expression and distribution
The regulatory effects of the modified NPs on PepT1 were investigated on protein and gene level. The hPepT1-Hela cells were seeded in six-well plate at a density of 1 × 105 cells/well. After 24 h of attachment, cells were treated with 5 μg/mL NSPV1000 NPs for 1, 2, 4, 8, 12, and 24 h. At each time point, cells were collected and the membrane and cytosol proteins were extracted using a membrane protein extraction kit (Beijing Dingguo Changsheng Biotechnology. Co., Ltd.). The segregated proteins were analyzed by western blot with the same method described above. Cadherin and β-Actin were used as membrane and cytosol control, respectively.
To further test the change of gene expression during the transporter-mediated endocytosis, mRNA was extracted from hPepT1-Hela cells using RNAiso Plus® Reagent Kit (Takara Biotechnology, Dalian, China), after being treated with 5 μg/mL NSPV1000 NPs for the same periods of time. Then the extracted mRNA was reversely transcribed to cDNA using PrimeScript® RT Reagent Kit with gDNA Eraser (Takara Biotechnology, Dalian, China). Quantitative real-time polymerase chain reaction (PCR) was conducted using SYBR Green PCR Master Mix, SYBR® Premix Ex TaqTM Kit (Takara Biotechnology, Dalian, China), and an ABI prim 7500 Sequence Detection System (Applied Biosystems, Foster City, CA). β-Actin was analyzed as control. The alteration of folds for hPepT1 gene expression relative to β-actin was calculated by 2−ΔΔCT method. The primer sequences of hPepT1 is forward (5’-3’): TGTCACGGGATTGGAATTCTC, reverse (5’-3’): TACTCGGCCCACTGTTTGCT, and for β-Actin is forward (5’-3’): CGCGAGAAATGACCCAGAT, and reverse (5’-3’): GTACGGCCAGAGGCGTACAG, respectively (synthesized by Invitrogen, Shanghai).
Animals
Male Sprague-Dawley rats weighing 220–250 g were purchased from Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). Animals were fasted 12 h with free access to water prior to the experiments. All the animal experiments in this study were performed under Institutional Animal Care and Use Committee-approved protocols of Shenyang Pharmaceutical University.
In situ single-pass intestinal perfusion study in rats
The in situ absorption performance was evaluated by single-pass intestinal perfusion method. Rats were anesthetized by intraperitoneal injection of 20% urethane solution (1 g/kg). The intestinal segments (about 10 cm) were exposed and segregated, including duodenum, jejunum, and ileum. After gently washed by saline, the intestinal segments were equilibrated by Krebs Ringer buffer (K-R buffer, containing 7.8 g NaCl, 0.35 g KCl, 1.37 g NaHCO3, 0.02 g MgCl2, 0.32 g NaH2PO4, 1.4 g glucose, and 0.32 g of CaCl2 in 1000 mL of purified water) and then were perfused with various 10 μg/mL DTX-loaded formulations diluted by K-R buffer. Gravimetric method was utilized in the calculation of the absorption rate (Ka) and apparent permeability (Papp). Samples were collected from the outflow at time intervals of 15, 30, 45, 60, 75, and 90 min, respectively. In the end, tested rats were sacrificed and the intestinal segments were excised and the length and radius of each perfused intestinal segment were recorded. The protein content of the samples was precipitated by adding three folds methanol followed by centrifuging for 10 min at 13,000 rpm. The processed samples were determined by HPLC. Ka and Papp of DTX solution and DTX-loaded formulation (unmodified NPs, NSPV1000 NPs, and NSPP1000 NPs) were calculated using the following EquationEquation (1)(1)
(1) :
(1)
(1)
Where Cin is the DTX concentration of the donor solution, Cout is the DTX concentration of the sample collected, and min and mout are weight change of the donor solution and the weight of the sample at each time interval, respectively. While r and l are the radius and length of each intestinal segment and v is the perfusion flow rate.
Biodistribution of the dipeptide-modified NPs in the gastrointestinal (GI) tract
The biodistribution of C6 solution, C6-unmodified NPs and C6-NSPV1000 NPs in GI tract were observed by CLSM (FluoViewFV 1000, Olympus, Japan). C6 formulations were administrated orally at a dosage of 5 mg/kg and rats were sacrificed after 45 min. Duodenum, jejunum, and ileum were excised, everted, and embedded in the optimum cutting temperature (OCT) embedding material and stored at −80 °C. Seven micrometer-thick slices were prepared and placed on cationic resinous slides and were later fixed with 4% PFA for 10 min. Triton X100 solution (1%) was used to improve the cell membrane permeability. After being rinsed with cold PBS carefully, the F-actin was stained with rhodamine phalloidin (red), and the nucleuses were stained with 4',6-diamidino-2-phenylindole (DAPI; blue). Subsequently, the slices were visualized under confocal laser microscopy (CLSM).
Pharmacokinetics evaluation
Male Sprague-Dawley rats were randomly divided into three groups (five rats per group) and were orally administrated with DTX solution (prepared similarly with Taxotere), DTX unmodified NPs, and DTX NSPV1000 NPs at a single DTX dosage of 10 mg/kg. Blood samples (approximately 0.3 mL) were withdrawn from retro-orbital sinus at different time points: 0.083, 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, and 24 h. Plasma samples were obtained by immediate centrifugation at 13,000 rpm for 10 min and stored at −20 °C.
Liquid-liquid extraction method was applied to process the serum samples with methyl tert-butyl ether as extraction agent. To 100 μL of plasma, 100 μL paclitaxel solution (internal standard, I.S.), 100 μL acetonitrile, and 3 mL methyl tert-butyl ether were added to extract DTX. The mixture was vortexed for 3 min, followed by centrifugation for 10 min at 3500 rpm. Then the supernatants were evaporated under nitrogen flow and the residues were dissolved in 100 μL of acetonitrile for analysis. The quantification of DTX in plasma was carried out using ultra performance liquid chromatography - tandem mass spectrometer (UPLC-MS/MS; TQD detector, Waters Corporation, Milford, MA). Optimum chromatographic separation was performed using a gradient of acetonitrile and water containing 0.1% formic acid on an ACQUITY UPLC BEH C18 column (50 mm ×2.1 mm, 1.7 mm). The ion transitions were monitored as follows: 830.4 → 304 for DTX and 854.5 → 286.3 for PTX. The pharmacokinetic parameters were calculated by the DAS software (Las Vegas, NV).
Statistical analysis
All the quantitative data were described using the mean ± SD (standard deviations), and statistical analysis was performed with Student’s t-test. p < .05 was considered statistically significant.
Results and discussions
Synthesis of dipeptide-modified PepT1-targeting polymer
Dipeptide (L-valine-valine, L-valine-phenylalanine) modified polyoxyethylene stearate with different PEG chain lengths (Mw: 1000, 2000) were synthesized by a facile approach (Supplementary Figure S1). Cbz-L-val-val and Cbz-L-val-phe were conjugated to the hydroxyl group of polyoxyethylene stearate. Then the obtained compounds were deprotected to expose the amino group, which is necessary for substrate recognition with PepT1 (Zhang et al., Citation2013b). The chemical structures of dipeptide-modified polymers were confirmed by 1H-NMR (Supplementary Figures S2–9), suggesting that the dipeptides were successfully coupled to polyoxyethylene stearate.
Preparation and characterization of NPs
The modified and the unmodified DTX-loaded NPs were prepared by emulsion-solvent evaporation method. According to the surface nitrogen analysis of the modified NPs with different dipeptide modified densities, the nitrogen content was detected to be 0.2 for 5% NSPV1000 NPs, 0.39 for 10% NSPV1000 NPs, and 0.44 for 20% NSPV1000 NPs (), indicating a saturated incorporation of the dipeptide-modified polymer with PLGA NPs. Hence, the optimal dipeptide modification percentage of 10% was applied in the fabrication of the modified NPs. All the modified NPs displayed an average size distribution around 165 nm, a comparable zeta potential ranging from −2.23 to −4.05 mV, and desirable drug loading capacity and encapsulation efficiency of DTX (Supplementary Table S1). The TEM image of NSPV1000 NPs is shown in , with the homogeneous spherical-shaped particle sizes in line with the DLS results. As shown in , various characteristic diffraction peaks of DTX from 4 to 27° were observed in free DTX and the physical mixtures, while the blank NPs and DTX-loaded NPs only showed a mild broad peak without appearance of the crystal peaks for DTX, suggesting the drug was encapsulated into NPs mainly in an amorphous or molecular form. In addition, NSPV1000 NPs exhibited good colloidal stability in various mediums for 24 h (), which was beneficial for oral delivery against the harsh GI environment. Besides, drug release performances were conducted using a dialysis method. As shown in , these nanoformulations displayed similarly sustained release without being influenced by the surface dipeptide decoration. Moreover, the NPs showed desirable sustained release in pH 1.0 (Supplementary Figure S11), indicating the survival of NPs from the harsh acidic conditions.
Figure 1. (A) Schematic illustration of the dipeptide-modified NPs; (B) Size distribution of NSPV1000 NPs; (C) Morphology of docetaxel-loaded NSPV1000 NPs by TEM.
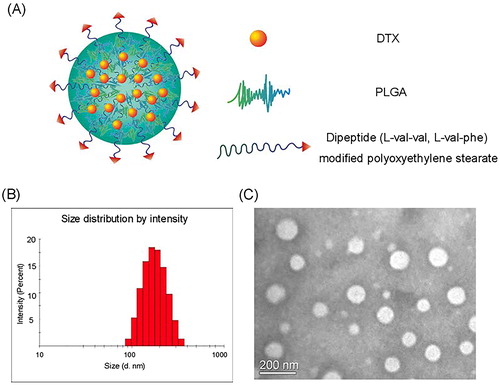
Figure 2. Characterizations of modified NPs. (A) X-ray photoelectron spectroscopy (XPS) curves of NPs with different surface densities of valine-valine decoration; (B) X-ray diffractions of DTX, blank NSPV1000 NPs, the physical mixture of DTX, blank NSPV1000 NPs, and DTX-loaded NSPV1000 NPs; (C) Colloidal stability of NSPV1000 NPs in the presence of different mediums at 37 °C for 24 h; (D) In vitro release of DTX solution, DTX-loaded unmodified NPs, NSPV1000 NPs, NSPV2000 NPs, NSPP1000 NPs, and NSPP2000 NPs in pH 6.8 medium for 24 h. (Data are shown as mean ± SD, n = 3).
Cellular PepT1 expression
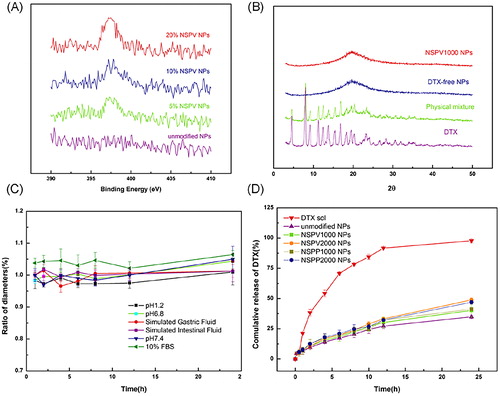
Stably transfected hPepT1-Hela cells were used to assess the delivery feasibility in vitro due to its specific overexpression of hPepT1 and mock-Hela with lower expression was utilized as a control. Western blot was used to determine the transporter expression levels. As shown in , specific bands with a molecular weight of 75 kDa representing the targeting protein were found in hPepT1-Hela cells, while hardly any signal could be observed in mock-Hela cell. The blotted data suggested the abundant expression of PepT1 in hPepT1-Hela cells that could serve as a suitable model for targeting efficiency analysis.
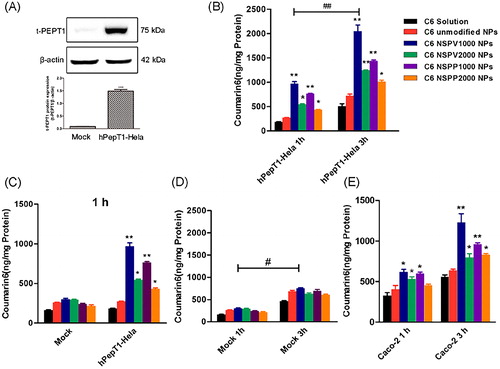
Figure 3. (A) Protein expression of PepT1 in stably transfected hPepT1-Hela and mock-Hela cell lines; Cellular uptake of C6 solution and C6-loaded unmodified NPs, NSPV1000 NPs, NSPV2000 NPs, NSPP1000 NPs, and NSPP2000 NPs in hPepT1-Hela cells for 1 and 3 h (B), in mock-Hela and hPepT1-Hela cells for 1 h each (C), in mock-Hela cells for 1 and 3 h (D), and in Caco-2 cells for 1 and 3h (E), respectively. Data are shown as mean ± SD. *p < .05, **p < .01 vs. cellular uptake of C6 solution group at the same time point, #p < .05, ##p < .01 vs. corresponding cellular uptake of each group at 1h, n = 3.
Cellular uptake
C6 was encapsulated into NPs to evaluate the cellular uptake efficiency. As shown in , C6 solution and C6-unmodified NPs showed similar cellular uptake in both mock, hPepT1-Hela cells. By contrast, the dipeptide modified C6-NPs demonstrated significantly higher uptake in PepT1 over-expressed hPepT1-Hela compared with those in mock-Hela, with a 3.76-fold higher for C6-NSPV1000 NPs, 2.02-fold higher for C6-NSPV2000 NPs, 3.26-fold higher for C6-NSPP1000 NPs, and 1.86-fold higher for C6-NSPP2000 NPs, respectively. In Caco-2 cells, the modified NPs showed significantly higher intracellular accumulation than those of C6 solution and unmodified NPs both at 1 and 3h. Among the modified NPs, NSPV1000 NPs showed best uptake efficacy with a 1.95-fold higher than the unmodified NPs and a 1.37-fold higher than NSPP1000 NPs after treated for 3h. These results suggested PepT1 transporter was involved in the enhanced cellular uptake of modified NPs. Besides, dipeptide modified NPs with shorter PEG chain had better cellular uptake, due to the less impediment of steric hindrance by the longer PEG chain. The L-valine-valine modified NPs might take advantage of its relatively smaller isopropyl group on the dipeptide side chain, which facilitated a more flexible approach to the binding domain of PepT1. Thus, the dipeptide-modified NPs with shorter PEG chain were selected for further evaluation.
PepT1-mediated endocytosis mechanisms
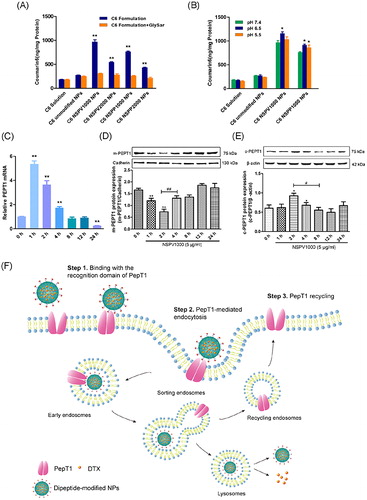
Mechanisms involved in the PepT1-mediated endocytosis were investigated in hPepT1-Hela and Caco-2 cells. A competitive inhibition experiment was carried out in the presence of GlySar (typical substrate of PepT1). In hPepT1-Hela cells, the uptake of all the dipeptide-modified NPs was substantially attenuated in the presence of GlySar. In contrast, the presence of GlySar had no significant influence on the uptake of C6 solution and C6-unmodified NPs. These results suggested that the competitive inhibitor GlySar would compete for the PepT1 binding pocket with the dipeptide exposed on the NPs surface, clearly demonstrating that PepT1 plays a critical role during the cellular uptake process. In addition, PepT1 is known for transporting small molecular substrates with the driven force of inward proton-gradient across the enterocyte membrane. Therefore, we further investigated the effect of proton on the cellular uptake of the modified C6-NPs. As shown in , a significantly higher internalization of the modified NPs was found when the extracellular pH was 6.5. By contrast, the cellular uptake of C6 solution and C6-unmodified NPs was independent on pH, indicating that the internalization of the modified NPs was a proton-dependent process. Similar data were generated in Caco-2 cells (Supplementary Figure S12). The morphology of cells in different acidic conditions was normal and intact observed under microscope after the experiment (Supplementary Figure S10).
Figure 4. (A) Competitive study in hPepT1-Hela cells in the presence of typical substrate GlySar (GS); (B) Influence of proton in the culture medium on the cellular uptake of dipeptide modified NPs in hPepT1-Hela cells; The variation of relative PepT1 mRNA expression versus β-actin (C) and the variations of membrane and cytosol PepT1 protein expression (D,E) after treatments with NSPV1000 NPs for different time over 24 h. Data are shown as mean ± SD. *p < .05, **p < .01 vs. C6/DTX solution group or control group, #p < .05, ##p < .01, n = 3; (F) The schematic illustration of hypothesized mechanism of PepT1-mediated endocytosis.
Regulatory effect of the modified NPs on PepT1
The influence of the dipeptide-modified NPs on the PepT1 expression and distribution of hPepT1-Hela cells was investigated. Cell samples were collected after being treated with 5 μg/mL NSPV1000 NPs for 1, 2, 4, 8, 12, and 24 h, respectively. To elucidate the spatial changes over the time, membrane and cytosol proteins were separated using an extraction kit. As shown in , the expression of membrane protein was significantly down-regulated after treatment with the dipeptide-modified NPs, following a gradual recovery to the normal level after 4 h of treatment. Meanwhile, the cytosol PepT1 protein expression showed the opposite trend, with significant increment after treatment for 2 and 4 h, and then decreased to a comparable expression with the untreated group. The mRNA expression was also assessed at same time-points, a dramatic mRNA expression increment of 5.6-fold was found after 1 h treatment and maintained at a relatively high level after 12 h of treatment. These results suggested that the surface transporter could serve as an anchoring site for the dipeptide-modified NPs and transport them into the cytoplasm, but the cytosol transporter could return to the cell membrane (Ding et al., Citation2017; Li et al., Citation2017; Tao et al., Citation2017b), and the hypothesized PepT1-mediated endocytosis is illustrated in . At the same time, a portion of new PepT1 protein might be synthesized as a supplement to maintain the normal level of membrane transporter.
In situ absorption
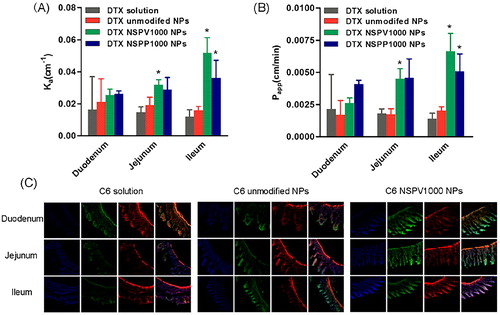
The intestinal absorption of the dipeptide-modified NPs was studied using in situ small intestinal perfusion. The absorption rate (Ka) and the apparent membrane permeability (Papp) of DTX solution, DTX-loaded unmodified NPs, NSPV1000 NPs, and NSPP1000 NPs in various intestinal segments are shown in . The modified NPs both had better absorption than those of DTX solution and unmodified NPs. NSPV1000 NPs had increased Ka of 1.20-, 2.16-, and 3.25-fold and increased Papp of 1.54-, 2.60-, and 3.28-fold in duodenum, jejunum, and ileum, compared to the unmodified NPs. While NSPP1000 NPs had increased Ka of 1.22-, 1.50-, and 3.06-fold and increased Papp of 2.4-, 2.65-, and 2.51-fold in duodenum, jejunum, and ileum than those of the unmodified NPs, respectively. Meanwhile, the DTX solution and unmodified NPs showed comparable absorption in all intestinal segments. Additionally, both the modified NPs showed highest absorption in ileum, in consistent with the previous report that ileum has the most abundant expression of PepT1 (Drozdzik et al., Citation2014). Therefore, the absorption of the modified NPs was relevant to the PepT1 abundance.
Figure 5. Intestinal absorption and distribution. (A, B) The in situ absorption rate (Ka) and the apparent permeability (Papp) of DTX solution, DTX-loaded unmodified NPs, NSPV1000 NPs, and NSPP1000 NPs. Data are shown as mean ± SD, n = 3. *p < .05, compared with unmodified NPs in each corresponding intestinal segment. (C)The gastrointestinal tract distribution of C6 solution, C6-loaded unmodified NPs, and NSPV1000 NPs 45 min after oral administration (Green). The nucleuses were stained with DAPI (blue), the F-actin was stained with rhodamine phalloidin (red) in the histological sections (magnification: ×200).
Biodistribution of C6-NPs in the GI tract
The biodistribution of different C6-NPs in the GI tract was evaluated by CLSM. Intestinal segments including duodenum, jejunum, and ileum were excised 45 min after oral administration. Nucleuses were stained with DAPI (blue) and F-actin was stained by rhodamine-phalloidin. As shown in , both the C6-unmodified NPs and C6-NSPV1000 NPs showed better accumulation than C6 solution in the enterocytes and C6-NSPV1000 NPs exhibited higher accumulation and permeation into the intestine villi in all intestinal segments when compared with C6 solution and the C6-unmodified NPs.
Oral absorption in vivo
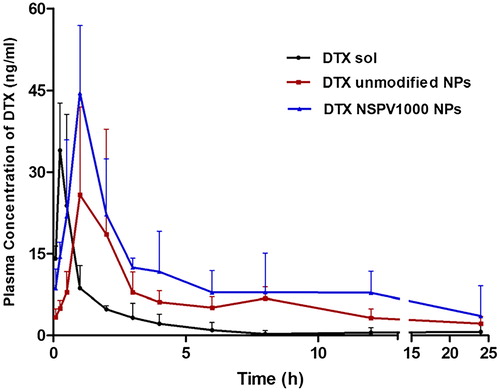
The in vivo pharmacokinetics of DTX solution, DTX-loaded unmodified NPs, and NSPV1000 NPs was studied in male SD rats. The main pharmacokinetic parameters are summarized in Supplementary Table S2 and the drug concentration-time curve is shown in . As shown in , DTX solution demonstrated rapid absorption and quick elimination after administration, resulting in a low bioavailability. Both the unmodified NPs and NSPV1000 NPs presented a sustained in vivo release with a 5.59-fold increase of t max. NSPV1000 NPs elicited the longest plasma halflife (t1/2) of drug with 5.16-fold to that of DTX solution and 2.02-fold to that of the unmodified NPs. Meanwhile, NSPV1000 NPs showed a higher Cmax which was 1.88-fold to that of unmodified NPs. Besides, the bioavailability of NSPV1000 NPs was 4.39- and 1.95-fold higher than that of DTX solution and unmodified NPs, respectively. The high-efficient oral absorption of NSPV1000 NPs indicated the contribution of PepT1-mediated enterocytes absorption of the dipeptide-modified NPs.
Figure 6. Mean plasma concentration-time curves of DTX in SD rats after oral administration of DTX solution and DTX-loaded unmodified NPs and NSPV1000 NPs at a dose of 10 mg/kg (mean ± SD, n = 5).
Conclusions
In this study, we successfully synthesized the conjugates of dipeptide (L-valine-valine, L-valine-phenylalanine) and polyoxyethylene stearate (PEG Mw: 1000, 2000) to functionalize PLGA NPs for PepT1-targeted oral delivery of DTX. The modified NPs displayed increased cellular uptake in stably transfected hPepT1-Hela cells when compared with the unmodified NPs, as well as in Caco-2 cells. Among them, L-valine-valine modified NPs with shorter PEG chain (Mw: 1000) exhibited higher targeting efficiency in vitro and in vivo. The dipeptide conjugates retained the binding affinity to PepT1 and contributed to the increased cellular uptake in a proton-dependent manner. More interestingly, the treatment of the modified NPs to cells could regulate the membrane and cytosol PepT1 protein expression and distribution, together with the alteration of the PepT1 mRNA transcription. It is the first time to elucidate the detailed self-maintenance mechanism of membrane PepT1 after being treated with the dipeptide-modified NPs. Therefore, the enhanced intestinal permeability and oral bioavailability of DTX indicated the promising application prospect of the dipeptide-modified NPs in oral delivery of various hydrophobic drugs with low oral bioavailability.
Supporting_information.doc
Download MS Word (2.5 MB)Acknowledgements
We would like to appreciate Prof. Kexin Liu from Dalian Medical University for providing stably transfected hPepT1-Hela cell line and mock-Hela cell line and all the research support.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- An S, Lu X, Zhao W, et al. (2016). Amino acid metabolism abnormity and microenvironment variation mediated targeting and controlled glioma chemotherapy. Small 12:5633–45.
- Ding L, Zhu X, Wang Y, et al. (2017). Intracellular fate of nanoparticles with polydopamine surface engineering and a novel strategy for exocytosis-inhibiting, lysosome impairment-based cancer therapy. Nano Lett 17:6790–801.
- Drozdzik M, Groer C, Penski J, et al. (2014). Protein abundance of clinically relevant multidrug transporters along the entire length of the human intestine. Mol Pharm 11:3547–55.
- Fan J, Li Z, Liu X, et al. (2018). Bacteria-mediated tumor therapy utilizing photothermally-controlled TNF-alpha expression via oral administration. Nano Lett 18:2373.
- Fang L, Wang M, Gou S, et al. (2014). Combination of amino acid/dipeptide with nitric oxide donating oleanolic acid derivatives as PepT1 targeting antitumor prodrugs. J Med Chem 57:1116–20.
- Ganapathy M, Huang W, Wang H, et al. (1998). Valacyclovir: a substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2. Biochem Biophys Res Commun 246:470–5.
- Guo X, Meng Q, Liu Q, et al. (2014). Simultaneous determination of three dipeptides (JBP485, Gly-Sar and JBP923) in the cell lysates by liquid chromatography-tandem mass spectrometry: application to identify the function of the PEPT1 transfected cell. Biomed Chromatogr 28:1839–45.
- Guo X, Meng Q, Liu Q, et al. (2012). Construction, identification and application of HeLa cells stably transfected with human PEPT1 and PEPT2. Peptides 34:395–403.
- Han X, Sun J, Wang Y, et al. (2015). PepT1, ASBT-linked prodrug strategy to improve oral bioavailability and tissue targeting distribution. Curr Drug Metab 16:71–83.
- Jiang X, Xin H, Ren Q, et al. (2014). Nanoparticles of 2-deoxy-D-glucose functionalized poly(ethylene glycol)-co-poly(trimethylene carbonate) for dual-targeted drug delivery in glioma treatment. Biomaterials 35:518–29.
- Kou L, Bhutia Y, Yao Q, et al. (2018). Transporter-guided delivery of nanoparticles to improve drug permeation across cellular barriers and drug exposure to selective cell types. Front Pharmacol 9:27.
- Kou L, Yao Q, Sivaprakasam S, et al. (2017). Dual targeting of l-carnitine-conjugated nanoparticles to OCTN2 and ATB(0,+) to deliver chemotherapeutic agents for colon cancer therapy. Drug Deliv 24:1338–49.
- Li L, Di X, Wu M, et al. (2017). Targeting tumor highly-expressed LAT1 transporter with amino acid-modified nanoparticles: toward a novel active targeting strategy in breast cancer therapy. Nanomedicine 13:987–98.
- Li L, Di X, Zhang S, et al. (2016). Large amino acid transporter 1 mediated glutamate modified docetaxel-loaded liposomes for glioma targeting. Colloids Surf B Biointerfaces 141:260–7.
- Li J, Guo Y, Kuang Y, et al. (2013). Choline transporter-targeting and co-delivery system for glioma therapy. Biomaterials 34:9142–8.
- Luo Q, Gong P, Sun M, et al. (2016). Transporter occluded-state conformation-induced endocytosis: amino acid transporter ATB(0,+)-mediated tumor targeting of liposomes for docetaxel delivery for hepatocarcinoma therapy. J Control Release 243:370–80.
- Pereira C, Araujo F, Granja P, et al. (2014). Targeting membrane transporters and receptors as a mean to optimize orally delivered biotechnological based drugs through nanoparticle delivery systems. Curr Pharm Biotechnol 15:650–8.
- Qu X, Zou Y, He C, et al. (2018). Improved intestinal absorption of paclitaxel by mixed micelles self-assembled from vitamin E succinate-based amphiphilic polymers and their transcellular transport mechanism and intracellular trafficking routes. Drug Deliv 25:210–25.
- Ramanathan S, Qiu B, Pooyan S, et al. (2001). Targeted PEG-based bioconjugates enhance the cellular uptake and transport of a HIV-1 TAT nonapeptide. J Control Release 77:199–212.
- Rosenblum D, Joshi N, Tao W, et al. (2018). Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun 9:1410.
- Shan W, Zhu X, Liu M, et al. (2015). Overcoming the diffusion barrier of mucus and absorption barrier of epithelium by self-assembled nanoparticles for oral delivery of insulin. ACS Nano 9:2345–56.
- Shan W, Zhu X, Tao W, et al. (2016). Enhanced oral delivery of protein drugs using zwitterion-functionalized nanoparticles to overcome both the diffusion and absorption barriers. ACS Appl Mater Interfaces 8:25444–53.
- Sugawara M, Huang W, Fei Y, et al. (2000). Transport of valganciclovir, a ganciclovir prodrug, via peptide transporters PEPT1 and PEPT2. J Pharm Sci 89:781–9.
- Sun Y, Sun J, Shi S, et al. (2009). Synthesis, transport and pharmacokinetics of 5'-amino acid ester prodrugs of 1-beta-D-arabinofuranosylcytosine. Mol Pharm 6:315–25.
- Tang L, Fu L, Zhu Z, et al. (2018). Modified mixed nanomicelles with collagen peptides enhanced oral absorption of Cucurbitacin B: preparation and evaluation. Drug Deliv 25:862–71.
- Tao W, Ji X, Xu X, et al. (2017a). Antimonene quantum dots: synthesis and application as near-infrared photothermal agents for effective cancer therapy. Angew Chem Int Ed Engl 56:11656–900.
- Tao W, Zeng X, Liu T, et al. (2013). Docetaxel-loaded nanoparticles based on star-shaped mannitol-core PLGA-TPGS diblock copolymer for breast cancer therapy. Acta Biomater 9:8910–20.
- Tao W, Zeng X, Wu J, et al. (2016). Polydopamine-based surface modification of novel nanoparticle-aptamer bioconjugates for in vivo breast cancer targeting and enhanced therapeutic effects. Theranostics 6:470–84.
- Tao W, Zhang J, Zeng X, et al. (2015). Blended nanoparticle system based on miscible structurally similar polymers: a safe, simple, targeted, and surprisingly high efficiency vehicle for cancer therapy. Adv Healthc Mater 4:1203–14.
- Tao W, Zhu X, Yu X, et al. (2017b). Black phosphorus nanosheets as a robust delivery platform for cancer theranostics. Adv Mater 29:1603276.
- Yan Z, Sun J, Chang Y, et al. (2011). Bifunctional peptidomimetic prodrugs of didanosine for improved intestinal permeability and enhanced acidic stability: synthesis, transepithelial transport, chemical stability and pharmacokinetics. Mol Pharm 8:319–29.
- Zhang Y, Sun J, Gao Y, et al. (2013a). A carrier-mediated prodrug approach to improve the oral absorption of antileukemic drug decitabine. Mol Pharm 10:3195–202.
- Zhang Y, Sun J, Sun Y, et al. (2013b). Prodrug design targeting intestinal PepT1 for improved oral absorption: design and performance. Curr Drug Metab 14:675–87.
- Zhu X, Wu J, Shan W, et al. (2016). Polymeric nanoparticles amenable to simultaneous installation of exterior targeting and interior therapeutic proteins. Angew Chem Int Ed Engl 55:3309–12.