
Abstract
Numerous papers analyze ground-level ozone (O3) trends since the 1980s, but few have linked O3 trends with observed changes in nitrogen oxide (NOx) and volatile organic compound (VOC) emissions and ambient concentrations. This analysis of emissions and ambient measurements examines this linkage across the United States on multiple spatial scales from continental to urban. O3 concentrations follow the general decreases in both NOx and VOC emissions and ambient concentrations of precursors (nitrogen dioxide, NO2; nonmethane organic compounds, NMOCs). Annual fourth-highest daily peak 8-hr average ozone and annual average or 98th percentile daily maximum hourly NO2 concentrations show a statistically significant (p < 0.05) linear fit whose slope is less than 1:1 and intercept is in the 30 to >50 ppbv range. This empirical relationship is consistent with current understanding of O3 photochemistry. The linear O3–NO2 relationships found from our multispatial scale analysis can be used to extrapolate the rate of change of O3 with projected NOx emission reductions, which suggests that future declines in annual fourth-highest daily average 8-hr maximum O3 concentrations are unlikely to reach 65 ppbv or lower everywhere in the next decade. Measurements do not indicate increased annual reduction rates in (high) O3 concentrations beyond the multidecadal precursor proportionality, since aggressive measures for NOx and VOC reduction are in place and have not produced an accelerated O3 reduction rate beyond that prior to the mid-2000s. Empirically estimated changes in O3 with emissions suggest that O3 is less sensitive to precursor reductions than is found by the CAMx (v. 6.1) photochemical model. Options for increasing the rate of O3 change are limited by photochemical factors, including the increase in NOx sensitivity with time (NMOC/NOx ratio increase), increase in O3 production efficiency at lower NOx concentrations (higher O3/NOy ratio), and the presence of natural NOx and NMOC precursors and background O3.
Implications: This analysis demonstrates empirical relations between O3 and precursors based on long term trends in U.S. locations. The results indicate that ground-level O3 concentrations have responded predictably to reductions in VOC and NOx since the 1980s. The analysis reveals linear relations between the highest O3 and NO2 concentrations. Extrapolation of the historic trends to the future with expected continued precursor reductions suggest that achieving the 2014 proposed reduction in the U.S. National Ambient Air Quality Standard to a level between 65 and 70 ppbv is unlikely within the next decade. Comparison of measurements with national results from a regulatory photochemical model, CAMx, v. 6.1, suggests that model predictions are more sensitive to emissions changes than the observations would support.
Introduction
Since the 1990s, a large number of tropospheric ozone (O3) trend analyses have been reported (e.g., Fiore et al., Citation1998; Chan, Citation2009; Lefohn et al., Citation2010; Oltmans et al., Citation2006, Citation2013; Cooper et al., Citation2012; Simon et al., Citation2015). These studies have focused on a range of spatial scales from global–intercontinental (e.g., Cooper et al., Citation2014) to urban (e.g., Los Angeles, CA: Pollack et al., Citation2013). The presence and production of O3 in the troposphere involve the complex chemistry of volatile organic compounds (VOC) and nitrogen oxides (NOx = NO + NO2), as well as carbon monoxide (CO) and methane (CH4). At scales of less than a few hundred kilometers, continental O3 chemistry at the ground depends mainly on VOC and NOx concentrations and the ratio of the two species. Since O3 is a regulated pollutant and emissions of its precursors are dominated by anthropogenic activity in most U.S. regions and population centers, controls of anthropogenic sources are of regulatory interest. Relatively few reports in the literature directly discuss NOx and VOC (or nonmethane organic compounds [NMOC], a measured subset of VOC) precursor trends in relation to O3 trends (e.g., Wolff et al., Citation2001; Butler et al., Citation2011). Perhaps most widely available are U.S. Environmental Protection Agency (EPA) (EPA, Citation2013a, Citation2014a) reports discussing national and regional O3 concentrations qualitatively in the light of precursor emission trends. Additional comparisons derive from examples of local or regional analyses in California, the Northeast, and the South: for example, the California Air Resource Board (CARB, Citation2014a), the New York Department of Environmental Conservation (Bureau of Air Quality Analysis and Surveillance, Citation2007; Department of Environmental Conservation [NYS], Citation2013), the Connecticut Department of Energy and Environmental Protection (DEEP, Citation2009), and the Texas Commission on Environmental Quality (TCEQ, Citation2013). Virtually no comparisons of long-term trends with air quality model projections since 1990 have been reported except in Hogrefe et al. (Citation2011) and Xing et al. (Citation2015).
Comparisons between precursor emissions, ambient precursor concentrations, and ambient O3 concentrations are important for estimating the effectiveness of precursor controls in reducing O3, even though there are limitations to these comparisons. Comparison of long-term trends provides observational confirmation of O3 response to precursor emission changes projected by air quality modeling. Rather than repeat the surveys of O3 trends per se, we contribute to improving the understanding of O3 trends in the continental United States by addressing the relationships between VOC and NOx emissions and ambient NMOC and NO2 concentrations relative to O3 changes. Currently there exist a limited number of concurrent sets of NOx and VOC emissions and both ambient precursor and O3 data to make comparisons of trends at specific locations (e.g., Blanchard et al., Citation2010a, Citation2010b, Citation2014a). Although there are a large number of sites for O3 measurement in the United States, the locations for measurement of NOx or NOy, total reactive nitrogen concentrations, are sparser; trend data for ambient NMOC are largely confined to the photochemical assessment measuring stations (PAMS) network comprising only 24 sites (Table S1).
In this paper, we examine associations of long-term emission trends with ambient precursor and O3 concentrations at multiple spatial scales, from continental to urban. These trends provide the basis for a quantitative analysis of O3 precursor relationships focusing on NO2 concentrations as a causal indicator for O3 production. A detailed emissions and ambient data analysis gives us a framework for projecting the O3 response to future precursor changes. Emission trends combined with the impact of regulatory initiatives are compared with photochemical model projections charting the influence of future emission changes on ambient O3 concentrations to 2025.
Methods
Retrospective analyses required that precursor emissions, ambient precursor concentrations, and O3 be measured consistently over a period of several years. To develop our comparisons, we adopted analyses from the literature with additional data analysis, assuming that (1) the data used are consistent with the reports from EPA’s national database, and (2) the data adopted for our investigation of regional trends are self-consistent over the study periods. Analyses are presented for periods from 1980 or 1990, depending on data availability, to 2013. Trends using various measures of O3 concentrations were examined, including the median and other percentiles of the cumulative distributions. Many trend analyses have focused on a regulatory measure, the daily peak 8-hr average O3 concentration, and the annual fourth-highest daily peak 8-hr average concentration averaged over a period of 3 years. The survey covers a number of different example locations over different time periods. In addition to aggregated national observations, we look at regional and urban conditions based on EPA’s (Citation2013b) designated climate-air quality regions shown in Figure S1. The urban locations in the survey are selected by region for regions that are in or near “nonattainment” of the current U.S. National Ambient Air Quality Standard (NAAQS) for O3.
The O3 NAAQS is presently 75 ppbv stated for a 3-year average of the annual fourth-highest daily peak 8-hr ozone concentration (EPA, Citation2015a). In 2014, EPA proposed a revision to a level of 65 to 70 ppbv with consideration of 60 ppbv (EPA, Citation2014a). Since all monitoring sites in a county or other geographical area must meet the O3 NAAQS, EPA and state agencies use the data from the individual monitoring site with the highest 3-year average of the fourth-highest daily peak 8-hr average ozone concentration within an area for air quality attainment designations.
Trends in annual precursor emissions were adopted from EPA’s National Emission Inventory (NEI) or from regional and state inventories embedded in the NEI. The ambient data reported in the literature are averages of O3 concentrations as noted earlier, combined with annual averages of ambient NO2 and NMOC concentrations, and daily maximum NO2 concentrations. These are acquired from the U.S. national database for air quality measurements. Air monitoring is conducted either by EPA or by state agencies reporting their observations to the national database for their trends assessment. Measurements are obtained using EPA’s established reference methods or equivalents, and quality control or assurance practices; uncertainties in measurement methods are documented in the literature (e.g., Parrish and Fehsenfeld, Citation2000; Demerjian, Citation2000; Hidy et al., Citation2011, chap. 10). We assume, in particular, that EPA’s nationally reported NO2 trend data represent consistent, EPA-approved measurements despite known interferences by some components of NOz (= NOy – NOx).
Historical and projected emission estimates were obtained from EPA (Citation2015b, Citation2015c, Citation2015d) and the California Air Resources Board (CARB) (Citation2015a, Citation2015b). Emission inventories include measured and calculated quantities that approximate actual emissions with varying degrees of accuracy. Changes in emission estimation procedures affect the comparability of historical emissions over long periods (multiple decades), as discussed later in the Results and Discussion sections.
Ambient air quality data for O3 and NO2 were obtained from EPA (Citation2015e, Citation2015f) for all regions and supplemented with data from CARB for California (Citation2015b, Citation2015c). The data include annual measures (e.g., maximum, annual fourth-highest daily 8-hr peak concentration, or mean concentration) for concentrations of various species (O3, NO2) at individual monitoring sites (EPA, Citation2015d).
Our analysis considers the O3 design value (DV) (highest 3-year average of the annual fourth-highest daily peak 8-hr average concentrations determined for all local sites where measurements exist), as well as trends in metrics pertaining to mid-range and low O3 concentrations (e.g., mean, 10th percentile at a selected DV monitoring site). The EPA and CARB metrics are derived from validated measurements, and accompanying completeness statistics (number of hours or days per year with measurements) are provided. While data are available for all individual monitoring sites, we summarize our analysis using core-based statistical areas (CBSAs). CBSAs are defined by the U.S. Bureau of the Census and include 374 metropolitan (at least one urban area with population exceeding 50,000) and 581 micropolitan (population between 10,000 and 50,000) statistical areas, covering approximately half the geographical area of the continental United States (U.S. Bureau of the Census, Citation2015). Twenty-four CBSAs were selected for detailed O3 trend analysis, including areas consistently experiencing the highest O3 concentrations (southern California, inland central California, and Houston, TX), additional areas with counties where EPA projects that maximum O3 concentrations will continue to exceed 70 ppbv under 2025 baseline inventory emissions (e.g., New York City metropolitan area; Dallas–Fort Worth, TX, and San Antonio, TX; and Milwaukee, WI), and areas where EPA projects that maximum O3 concentrations will fall below 70 ppbv with implementation of 2025 baseline emissions (Atlanta, GA; Birmingham, AL; Cincinnati, Columbus, and Cleveland, OH; Chicago, IL; Detroit, MI; Memphis, TN) (EPA, Citation2014a). The 24 CBSAs selected for in-depth analysis include three CBSAs within each of eight regions having one or more O3 nonattainment designations (northeastern United States, southeastern United States, Texas, Ohio, southern Great Lakes, southern California, inland central California, San Francisco Bay area). If a selected CBSA lacked consistent, long-term data for O3 precursors, results are also reported for an alternate CBSA within the same region. Our study set includes a range of locations experiencing high O3 concentrations, which we supplement with additional CBSA and national analyses as noted. CBSAs are comprised of multiple complete counties, so the NO2 and O3 monitoring sites within each CBSA represent a range of urban and nonurban sites.
Two NAAQS apply to NO2: (1) the annual average of all hourly measurements (53 ppbv), and (2) the 3-year average of the 98th percentile of the daily peak 1-hr NO2, 100 ppbv (EPA, Citation2015a). All monitoring sites must meet these standards. We also report other NO2 metrics, such as the annual average of the daily peak 1-hr NO2 concentration. In addition to the maximum site, we average NO2 measurements across sites. Site data are weighted by the number of valid sampling days to generate intersite averages within CBSAs. The NO2 metrics are highly correlated: Correlations of the annual average of the daily peak 1-hr NO2 concentration with the annual average of all hourly measurements were r2 = 0.96 (maximum site data) and r2 = 0.88 (intersite means). The correlations of the maximum site NO2 metrics with intersite means ranged from r2 = 0.69 to 0.76. There is no NAAQS for VOC or NMOC, but there are emission constraints for species of VOC that are identified as toxins.
Ambient speciated NMOC observations were accessed through the EPA database (EPA, Citation2014b), largely representing PAMS data. PAMS stations are few in number relative to O3 and NO2, and are located mainly in or around areas of O3 nonattainment, listed in Table S1. The stations are not representative of the designated regions in Figure S1, but are assumed to be at least coarse indicators of urban or nonurban conditions in designated regions. The PAMS data generally are taken continuously in the summer months, and by canister sampling throughout the year.
Potentially predictive empirical relationships emerge from the emissions and ambient data comparisons, which we compare with air quality modeling results for future conditions across the United States. An example of photochemical model (EPA-CAMx, v. 6.1) calculations is used for this comparison. The model results have been reported by EPA for meeting a proposed revision of the O3 NAAQS with projected national and regional emission reductions (e.g., EPA, Citation2014a).
Results
Precursor emissions
National trends in NOx and VOC emissions derive from the National Emissions Inventory (NEI) (EPA, Citation2015a). Since 1990, the inventory has been developed from state inputs every 3 years, with intervening updates. EPA has projected emissions into the future using current regulatory initiatives combined with model estimates of emissions from different sectors of local infrastructure and the economy. As the inventory has evolved, there have been changes in the methods for estimation. Most of these are documented in the reports describing the inventory each 3 years (e.g., EPA, Citation2013a).
Annual extensions of the NEI have been reported (Xing et al., Citation2013; Blanchard et al., Citation2014b). The national trends for the precursor emissions since 1970 and projected to 2025 are shown in (EPA, Citation2015a, Citation2015b, Citation2015c). The EPA’s historical trend inventory is complicated by changes in emission estimation models and methods, especially for mobile sources, so the actual declines between 1980 and 2013 may have varied somewhat from those listed in . The estimated VOC emissions decline steadily until 2000, then tended to level off at 15–17 million tons (mT) per year (yr). In contrast, estimated NOx emissions were roughly constant until 2002–2003 at 27 mT/yr, when they decline steeply through 2013 to <10 mT/yr. Both NOx and the VOC emissions tend to level out after 2015 because no emission reductions beyond current regulatory initiatives are projected.
Table 1. U.S. national emissions of NOx and VOC (EPA, Citation2015a, Citation2015b, Citation2015c).
EPA’s current emission trend inventory shows that annual national level anthropogenic emissions declined from ~27 mT of NOx and ~35 mT of VOC in 1970 to ~13 mT NOx and ~18 mT VOC in 2013 (). NOx emissions from the 1980s may be underestimated (N. Kumar, personal communication), indicating larger actual declines. Some investigators have suggested that mobile source NOx emissions are overestimated after EPA began using MOVES in place of MOBILE6 (see ). For example, Fujita et al. (Citation2012) found that mobile source NOx emissions from the MOVES model overestimated NOx emissions that had been determined from measurements made in a highway tunnel. A fuel-based emission inventory by McDonald et al. (Citation2012) yielded relatively constant on-road mobile source NOx emissions from 1990 to 2000 (compared with a 12% reduction in EPA’s trend inventory), but comparable (within 7%) mobile source NOx emissions in 1999 and 2008. Xing et al. (Citation2013) report national NOx emissions of ~28 mT in 1990 and ~14 mT in 2010, compared with EPA’s trends inventory of 26 mT and 15 mT in 1990 and 2010, respectively, as shown in ; total NOx emission reductions between 1990 and 2010 are 48% according to Xing et al. (Citation2013) and 42% according to EPA (Citation2015d).
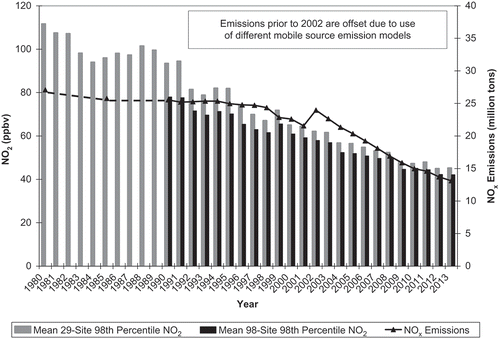
Figure 1. U.S. national NOx emissions compared with trends in 29-site and 98-site averages of the annual 98th percentile concentration of daily 1-hr maximum NO2 concentrations (EPA, Citation2015d, Citation2015e). Fewer trend sites operated continuously from 1980 (29) than from 1990 (98).
EPA projects an additional 4.6 mT decrease in NOx emissions and a 2.6 mT decrease in VOC emissions between 2013 and 2025 (). On-road mobile sources were the largest source of anthropogenic NOx and VOC emissions between 1970 and 2013 and prior to 2002, respectively. In 2013, the principal NOx emission sources were on-road mobile sources (5 mT), nonroad mobile sources (2.7 mT), electric utilities (1.8 mT), industrial and other fuel combustion (1.3 and 0.6 million tons, respectively), and industrial processes (1.2 mT) (EPA, Citation2015d).
Ambient concentrations
National trends for NO2
The U.S. national decline in annual average NO2 concentrations compared with NOx emissions shows an approximate linear 1:1 proportional relationship between emissions and ambient concentrations (). EPA reports a 60% decrease obtained from a 29-site average of the annual 98th percentile concentration of daily 1-hr maximum NO2 concentrations between 1980 and 2013 (this trend statistic reflects one of two National Ambient Air Quality Standards [NAAQS] for NO2; EPA selected sites based on continuity of operation) (EPA, Citation2015e). Corresponding declines were 46% between 1990 and 2013 based on 98 sites and 29% between 2000 and 2013 based on 180 sites (EPA, Citation2015e). These decreases are about 20% per decade. The 1980–2013 ambient NO2 trends are consistent with EPA historical NOx emission trends, although the ambient trends show a more consistent and nearly linear decline over time (). The relatively small discrepancy between ambient NO2 and NOx emission trends may reflect changes in emission estimation methods. Regardless of inconsistencies or inaccuracies in the emission inventories, the ambient NO2 measurements indicate a steady and sustained decrease in the ambient concentrations of NO2, consistent with NOx emission trends.
National trends for O3
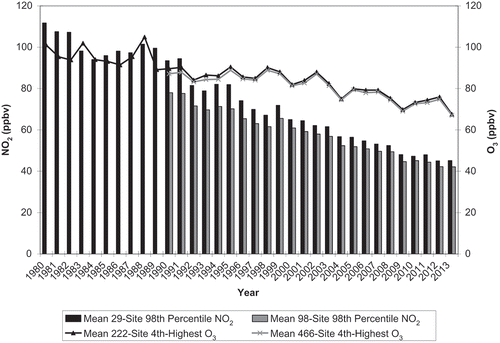
Averaged across either 222 O3 trend sites (1980–2013) or 466 O3 trend sites (1990–2013), the mean annual fourth-highest daily peak 8-hr O3 concentrations declined less rapidly than did the average ambient 98th percentile NO2 concentrations, averaged across either 29 NO2 trend sites between 1980 and 2013 or 98 NO2 trend sites between 1990 and 2013 (). As expected, the reported O3 concentration decreases occurred during a period of declining anthropogenic VOC and NOx emissions.
Figure 2. U.S. national trends in average annual 98th percentile concentration of daily 1-hr maximum NO2 concentrations compared with trends in average annual fourth-highest daily peak 8-hr O3 (EPA, Citation2015e; Citation2015f). Fewer sites operated from 1980 (29 for NO2 and 222 for O3) than from 1990 (98 for NO2 and 466 for O3).
Ozone trends over different spatial scales in the United States embody not only emissions and photochemistry, but seasonal meteorological features, including temperature, humidity, solar radiation, vertical mixing, and air mass transport. The national data represent an aggregate of measurements taken across the continent, averaging our continental meteorological features. Regional (Figure S1) analyses disaggregate observations mainly by geographical–climatological conditions adopted by EPA (Citation2013b). Urban or local analyses are represented by metropolitan or metropolitan-influenced areas where O3 concentrations are high compared with distant rural areas.
Regional-scale considerations
Decreasing O3 trends in the continental United States generally occur at the upper end of the cumulative O3 concentration distribution, in summer, and in less urbanized areas or at rural sites. More modest O3 trends generally occur at the lower end of the ozone distribution, often in the winter, and in more urbanized areas (Chan, Citation2009; Lefohn et al., Citation2010; Butler et al., Citation2011; Cooper et al., Citation2012; Simon et al., Citation2015). Some values of O3 decrease along with NOx and VOC emission reductions, as reported by the Midwest Ozone Group (Citation2013), and shown by region in Supplement Table S2. Even though most areas have indicated decreasing high-percentile O3 concentration trends, there are still a few locations with positive (increasing) trends reported in the continental United States (e.g., Simon et al., Citation2015; Table S2) and west or east coast of southern Canada (Chan, Citation2009).
In most locations, high (>95th percentile) and the fourth-highest daily 8-hr peak O3 average concentrations have decreased with declining NOx and NMOC emissions since the 1990s, mean daily 8-hr peak O3 concentrations have not decreased as much, and 5th–10th percentile concentrations have not decreased or have tended to increase, as indicated in (also Chan, Citation2009; Butler et al., Citation2011; Simon et al., Citation2015). The decline in high-percentile daily 8-hr peak O3 concentration after 1999, for example, generally ranges from 0 to 2%/yr, or about 0.2–1.5 ppbv/yr, in the United States and much of inland Canada. Consistent with previous studies, trends indicate that regional O3 concentrations are decreasing; these decreases are most pronounced at the highest O3 percentiles (averaging 1.1 to 1.3%/yr at the 95th through 99th percentiles), less pronounced at the 90th and 75th percentiles (averaging 0.9 and 0.6%/yr, respectively), and least pronounced or upward at the 50th and 10th percentiles (averaging –0.3%/ yr and +0.3%/yr, respectively) (). Trends in are expressed as percent per year for all metrics for comparability; downward trends are even more pronounced at higher percentiles when expressed in absolute terms (ppbv/yr). Possible explanations for increasing O3 trends at low (5th–10th) percentiles include increasing background O3 concentrations, potentially associated with intracontinental transport or intercontinental transport across the Pacific Ocean and affecting western North America (Cooper et al., Citation2012). Atmospheric chemical reactions may also contribute to increasing O3 concentrations at low percentiles, especially reduced titration of O3 by NO, as NOx emissions have declined (Lefohn et al., Citation2010; Cooper et al., Citation2012).
Table 2. Trends in daily peak 8-hr O3 by Core Based Statistical Area (CBSA) and metric, 1990–2014.
The variability of observed trends in O3 concentrations across seasons and percentiles suggests that the relationships between O3 and its precursors, VOC and NOx, vary across space, time, and meteorological conditions. Year-to-year variations in maximum O3 concentrations are known to result from changes in weather, in which some years have more days with conditions favoring O3 formation and accumulation (higher temperatures, sunlight, or low wind speeds). Weather-adjusted O3 trends dampen the year-to-year variation, making trends more apparent over shorter time periods without otherwise altering the overall multidecadal downward trends (EPA, Citation2015f; also Figure S2).
Since O3 at ground level has a short lifetime of approximately 1 day, spatial scale considerations <~500 km are more relevant to characterizing concentration trends in this gas than the national levels. Climatic regions in the United States have been adopted as indicated in Figure S1 by EPA (Citation2013b). Ozone-precursor relationships can differ among regions. In general, ambient precursor data availability is limited within regions of the United States, with certain ambiguities associated with the NO2 and NMOC measurements (e.g., Demerjian, Citation2000; Parrish and Fehsenfeld, Citation2000; Hidy et al. Citation2011, chap. 10). Thus, much of the previous trend assessment on a regional or smaller scale has relied on comparison of O3 changes with precursor emissions rather with ambient data. Regional differences and similarities are well illustrated by comparing annual NEI NOx and NMOC emissions with mean summer O3 concentrations from 2000 to 2012. These results are illustrated in Figure S2 for the meteorologically adjusted mean summer peak 8-hr O3 concentrations and the precursor emissions in EPA’s nine designated climatic regions. The adjustment method is specified by Camalier et al. (Citation2007). Inspection of these trends suggests that summer mean peak 8-hr O3 concentrations have generally declined in most of the designated regions (except the Southwest) at a regional average rate of ~1%/yr between 2000 and 2012. This is parallel with annual average NO2 reductions of ~3%/yr and VOC (for 6 regions) of ~2.5%/yr. (The Southwest, South, and West North Central regions show positive VOC emissions). The O3 design value (DV) results are qualitatively consistent with the reduction estimates by region from the Midwest Ozone Group (Citation2013) shown in Table S2. Some of the regional mean O3 trends in Figure S2 suggest a potential increase from 2010 to 2012, or at least a concentration leveling. It is uncertain whether this is an anomaly at the terminus of the sequence, or “real” occurrence in the sequence of annual trends. In the central and northeast regions (Figure S2), a decline is suggested with the implementation of the Ozone Transport NOx State Implementation Plan strategy, NOx SIP Call (EPA, Citation1998; EPA, Citation2005), after 2002, but the shift in the mean, meteorologically adjusted change does not deviate substantially from an overall decline between 2000 and 2012. The decrease in O3 DV reported by the Midwest Ozone Group (Citation2013) in Table S2 follows the general decline in annual precursor emissions, noting that the statewide NEI-based annual emission trends in this work show a bump in levels in the mid-2000s that we attribute to an NEI change in motor vehicle emissions modeling rather than to a “real” change (e.g., Blanchard et al., Citation2014b). These results are qualified in the Butler et al. (Citation2011) O3–NOx emission comparison in the eastern United States in the two periods 1997–1999 and 2006–2008. Their results suggest that the NOx SIP Call contributed strongly to O3 reductions during the summer in eastern regions after 2002. Butler et al. qualify their results, noting that NOx and NMOC from motor vehicles or other sector emissions are also a likely factor, particularly in urban conditions.
Toward the urban scale
Interpretation of the trends in O3 chemistry is facilitated by examining conditions on a scale paralleling the lifetime of ground-level O3 and its precursors—hours to a day. Taking account of typical vertical mixing and air mass transport times, the spatial scale of interest is on the order of 100 to 500 km. The conditions for high O3 concentrations most frequently are associated with intensified photochemistry in large metropolitan areas either within their boundaries or immediately downwind of their population centers. Of particular interest in the United States are areas designated as nonattainment of the O3 NAAQS. Those listed in were designated in 1996 for nonattainment (see also Table S1). We summarize briefly the status of O3 and precursor trends in these locations. The nonattainment areas listed in represent important differences in climate and in conditions associated with multiscale impacts from surrounding source areas. These cover the megacity regime in the Northeast, and the megacity region around the Great Lakes. Additionally, major urban areas are listed along the coast of the Gulf of Mexico and in the Southeast, the southern border region of the arid Southwest, and California. Given in Table S3 are some conventional measures of O3-climate differences that are useful to keep in mind for this discussion. The climate conditions in Table S3 indicate a midsummer high daytime temperature of 67–96ºF and summer afternoon relative humidity of 28–68% across metropolitan locations. Annual precipitation increases from west to east, with sunlight pervasiveness strongest in California. Maximum summer mixing heights vary widely with the lower values along the coasts. Air mass transport conditions vary widely, with the Western locations much less susceptible to intracontinental transport than the Midwestern and Eastern sites. A measure of interstate impact of O3 on the locations in Table S3 is included, and shows the regions of the Great Lakes, the Northeast, and the Southeast are potentially most susceptible to interstate influence. Note that U.S. international border transport enters from southern Canada in the East Central region and the Northeast, and from northern Mexico for the Southwest (Hidy et al., Citation2011). Along the West Coast (e.g., Hidy et al., Citation2011), there is a known potential for intercontinental impact of O3 from transport across the Pacific Ocean (e.g., Oltmans et al., Citation2010; Parrish et al., Citation2009). The eastern Gulf Coast experiences a potential for intercontinental transport across the Atlantic Ocean from the African continent. There is substantial literature based on monitoring in these locations. We summarize information in several locations as examples of O3-precursor conditions on urban (~100 km) spatial scales.
The Northeast is exemplified by southern New York (dominated by the New York City metropolitan area), southern Connecticut, and the Baltimore–Washington, DC, area. The annual fourth-highest peak 8-hr O3 concentrations in New York declined 12–17% between 1980 and 2010. In contrast with the large (>50%) reduction in NOx and VOC emissions and similar decreases in NO2 and NMOC concentrations, the statewide average of the annual fourth-highest daily peak 8-hr O3 concentrations declined from ~85 ppbv in 1994–1996 to ~ 75 ppbv in 2010–2012 (a decrease of 12%) (DEC NYS, Citation2013; Blanchard and Hidy, Citation2013). At New York metropolitan area sites, the 1990–2003 daily peak 8-hr average O3 concentration ranged from 91 to 99 ppbv in 1999–2003, decreasing to 75–86 ppbv from 2004 to 2011 (decrease of 12–17%) (e.g., DEC NYS, Citation2013). In southwestern Connecticut, the 8-hr O3 DV decreased from ~120 ppbv between 1985 and 1990 compared with 90–100 in 2000–2005 (or ~18–25%). This is compared with monthly median NMOC summer declines of ~20–40 ppbv between 1996 and 2006, respectively, at Westford and East Hartford (~30–40%); NOx concentrations are reported declining from ~20 to ~12 ppbv at these sites (~40% reduction) (e.g., Connecticut DEEP, Citation2009).
Various studies (e.g., Gego et al, Citation2007; Gilliland et al., Citation2008; Godowitch et al., Citation2008; Aleksic et al., Citation2013; Zhou, et al., Citation2013) have attributed O3 reductions downwind (east) of the Midwest after 2002 to the influence of NOx reductions from the NOx SIP call (EPA, Citation1998, Citation2005). Yet the state-reported New York and southern Connecticut O3 decrease shows little or no discernible change with precursor emissions in the post-1997 period. Moreover, a comparable annual NO2 concentration reduction in New York after 2000 exhibits little apparent effect on the post-2000 O3 concentrations (see also Blanchard and Hidy, Citation2013). The daily peak 8-hr average O3 concentrations in New York or in southern Connecticut suggest only a weak, meteorologically masked long-term response in the rate of O3 decline with introduction of NOx reductions following the NOx SIP call implementation after 2002. This result is contrasted with the Aleksic et al. (Citation2013) eight-site analysis of O3 concentrations and ambient temperature for 1995–2002 versus 2003–2012. They deduced that maximum O3 concentrations decreased despite warmer midday conditions in later years. This O3 reduction was cited as evidence for the effect of the NOx SIP call.
The Baltimore, MD–Washington, DC, area is included in nonattainment status. Ozone in these cities responds to not only high-density local precursor emissions, but also westerly transport of pollution from the interior (Bergin et al., Citation2007; He et al., Citation2013). Ozone in these cities has steadily declined in concentration, ~0.62 ppbv/yr between 1997 and 2010 (~0.7%/yr), while NOx decreased from 65 to ~30 ppbv (~54% or 4%/yr). Most of the NOx decrease has come after 2003 with the NOx SIP call. The local and upwind NOx decrease from electric utility emission controls evidently does not parallel a corresponding proportional O3 change in the Northeast and Mid-Atlantic areas. Since CO is roughly proportional to NMOC concentrations in urban areas, the former is potentially an indicator of NMOC decline (e.g., Harley et al., Citation2004). In the Baltimore–Washington, DC, region this correspondence implies a NMOC decline of 40% in 13 years or ~3%/yr, similar to NOx decrease. As elsewhere, O3 trends in the Baltimore–Washington, DC, area depend on meteorological conditions. Walsh et al. (Citation2008) used a statistical cluster analysis of aerometric variables to show that O3 depends strongly on warm temperature, weak winds, and sunlight relative to other groups of variables analyzed. They conclude that (precursor-driven) changes in local chemistry were important for declines relative to the influence of air mass transport and other meteorological variables.
A second major urban-industrial region exists around the southern Great Lakes, including Detroit, MI, and Chicago, IL. A Southern Michigan Council of Governments (SEMCOG, Citation2010) analysis of trends for design values from 1992 through 2010 at nine sites in the Lake Michigan region shows no decline until 2002–2004; after this time a sharp decline in this measure is observed from ~85 to 66 ppbv. Koerber (Citation2010) reported a decline in DV for O3 in this region from 1991 to 2009 (~ 0.8%/yr), compared with 1990–2008 NOx and VOC reductions of ~22% and 41%, respectively. The EPA (Citation2014a) report for Detroit shows essentially no O3 trend from 1984 to 2005, with a decrease in annual average NO2 concentrations from 30 ppbv to 17 ppbv. Jing et al. (Citation2014) indicate a declining trend in May–August area-wide average daily 8-hr peak O3 concentrations for 2005-2009 in Chicago followed by an increasing trend in O3 maximum for 2009–2013 despite a reduction in summertime NO2 emissions of ~42% and NO2 average concentrations of ~25%. In Chicago, ambient NMOC exemplified by benzene, toluene, and oxygenates (e.g., formaldehyde and acetaldehyde) increased after 2009. Based on statistical analysis of daily ambient concentrations of O3, NO2, and NMOC between 2005 and 2008 compared with 2009 and 2013, Jing et al. (Citation2014) concluded that Chicago may have shifted from an NOx-sensitive chemical regime to an NMOC-sensitive regime about 2008–2009.
A third regional group includes cities representing the southern tier of the United States, including Houston, Dallas, and El Paso, TX, and Baton Rouge, LA. These cities are at or close to nonattainment based on the current O3 NAAQS, but represent distinctly different meteorological and emission regimes. The Houston area is a petroleum industry and harbor-based city growing in population and mobile sources. Dallas–Fort Worth is the second major industrial–commercial city complex with growing population in Texas, inland from Houston. El Paso is a moderate-sized commercial city in arid conditions at the northern border of Mexico. And Baton Rouge is a moderate-sized petroleum-processing-oriented commercial city, the capitol of Louisiana. For example, the Houston area has seen a substantial reduction in 3-year averages of the fourth- highest daily 8-hr peak O3 concentration, from 117 ppbv in 2000 to 90 ppbv in 2011 (TCEQ, Citation2013). Analysis of Sather and Cavendar (Citation2007, Citation2012) gives a perspective on ambient precursor to O3 conditions in these four cities from 1986 to 2010. Emissions of NOx and NMOC decreased in the cities during the period of study. For Houston, their trends in 3-year averages of the fourth- highest daily peak 8-hr O3 concentrations over this span of years declines at ~2%/yr. They report the Houston reduction in June–August 0500–0800 hourly local standard time (LST) NOx concentrations during the same period as ~42%. For June–August 1997–2000, weekday 0500–0800 hourly LST NMOC decreased ~5%. For Dallas–Fort Worth, the 3-year average of the fourth-highest daily peak 8-hr O3 concentrations declined from 100 ppbv in 1986 to 86 ppbv in 2010 (14% or ~1%/yr). Mean NO2 concentrations at three Dallas–Fort Worth sites declined about 46% between 1985 and 2006. For 1986–2010, NMOC concentrations decreased 39% based on one site. For El Paso, the 3-year average of the fourth-highest daily peak 8-hr O3 concentrations declined from 93 ppbv in 1986 to 71 ppbv in 2010 (24% or ~2%/yr). Annual mean NO2 concentrations decreased 44% between 1991 and 2010, and NMOC decreased by 65%. Baton Rouge shows a change in the average of the fourth-highest daily peak 8-hr O3 concentrations, which decreased from 1986 to 2010 by 22% or ~1.5%/yr. This is compared with reduction in mean NO2 concentration of 30% between 1996 and 2010, and 0600–0900 3-hour integrated NMOC concentration declines of 68%. In a separate analysis, Klasinc et al. (Citation2008) show an increasing trend in 12-month running average O3 between 1995 and 2005 for three sites in Baton Rouge.
The fourth regional group consists of a southeastern region from Atlanta, GA, and Birmingham, AL, to the coast of the Gulf of Mexico. These two cities currently represent near-nonattainment areas. This region exemplifies a post-World War II rapidly growing industrial–commercial region, where a semitropical climate exists with a broad span of vegetation and high natural VOC emissions. During the period between 1999 and 2013, NEI emissions data have been compiled and extended along with a series of O3 and precursor measurements in these cities and in the surrounding region (e.g., Hidy et al., Citation2014). Fractional annual emission reductions over this period for NOx were estimated to be 57% and for anthropogenic VOC 32% (excluding open burning). Anthropogenic NMOC superimposed on a high natural VOC baseline declined with emission controls over the sampling period. Statistically significant (p < 0.05) linear relationships between annual regional NOx emissions and ambient NOy and between VOC emissions and ambient NMOC concentrations in Atlanta and Birmingham are listed in Table S4. A rural–urban O3 comparison is shown in Table S4, indicating a declining trend in O3 concentration both in the region and in the two cities. Reduction in O3 concentrations followed the NOy decline and (Atlanta) NMOC decrease, but not with a 1:1 proportionality (Table S4). The NMOC relationship is ambiguous here because of the large contribution to total NMOC from natural sources. In any case, the statistical linearity of annual O3 response to the decline in NOx emissions in the Southeast after 2003 is consistent with other results regarding the impact of the NOx SIP call in the eastern United States.
Our fifth group is in EPA’s western region, mainly represented by urban or highly populated areas of California. This region includes major cities, Los Angeles and San Francisco, and the large agricultural and petroleum production-influenced San Joaquin and Sacramento Valley subregions of the state. California covers a huge area north of western Mexico and eastward from the Pacific Coast inland into the arid regimes of the Southwest. Pollution from activities in an environment of growing population, industry, and motor vehicle transportation since the 1940s is among the most severe experienced in the United States. Los Angeles has documented periods of intense smog since the 1950s. Its long record of O3 measurements in urban North America, dating back to this time, reported events with midday O3 concentrations exceeding maximum hourly averages of 600 ppbv. A linear decline in O3 as measured annually from 1979 to 2013 for the 3-year moving average of high percentile of the daily peak 8-hr average O3 concentration is 56% (1.7%/yr) in Los Angeles, 30% (0.9%/yr) in San Francisco, and 17% (0.5%/yr) in the San Joaquin Valley based on CARB data. In an alternative analysis, Pollack et al. (Citation2013) found the decline in 8-hr maximum O3 concentrations with average concentrations of precursors is fitted better with an exponential decrease compared with a linear one. The decreasing trend in the South Coast Air Basin (SoCAB) is steeper than in the other three airsheds, as indicated in Figure S3. The difference in O3 reduction rate is attributed in part to the sustained efforts of the state, and especially the South Coast Air Quality Management District (SCAQMD), reducing NO2 emissions by 62% (2%/yr) and annual reactive organic gases (ROG, analogous to NMOC) emissions by 78% (2%/yr) between 1979 and 2013. Pacific coastal aerometric conditions are also a factor. Decreasing VOC emissions slow the rate of O3 formation, taking longer to reach high O3 concentrations. With slower rates of O3 formation, the afternoon sea breeze can sweep air from the high emission density areas of the central Los Angeles Basin before O3 can reach high levels there, though O3 levels may still reach high concentrations further inland (Midwest Ozone Group, Citation2013).
The California O3 experience shows its complexity in trends found in the four adjacent air basins. The complexity is related in part to emission characteristics and trends, as well as conditions involving mesoscale mixing of the sea and land breeze as seen in the SoCAB and to an extent in the San Francisco Basin (SFOB). Coastal areas contrast with inland areas with respect to local chemistry combined with mesoscale transport and mixing due to prevailing westerly airflow from the Pacific Coast into the interior.
In summary, this national to near-urban survey indicates that measures of O3 concentrations have declined in the United States since the 1980s or 1990s, with both NOx and NMOC emissions decreasing nationally and in most locations. High percentiles of daily peak 8-hr average O3 concentrations typically have declined by 0.5–1.0%/yr in most of the United States after the mid 1990s. There are a few exceptions where O3 trends have tended to increase; these are located primarily in the western United States and in southern Canada. Where annual collections of daily data are complete and accessible, the O3 decline has taken place at a rate with a linear fit less than 1:1 proportional to either NOx or NMOC reduction. A notable exception is the case of the Los Angeles area, whose O3 and precursor decline fits better with an exponential decline, and the O3 decrease is similar to that of NOx emissions.
The role of NOx
In this section, we concentrate on the relationship between ambient O3 concentrations and nitrogen oxides, noting the evidence that NOx emissions and ambient NO2 track one another. The national trend for ambient NO2 concentrations generally tracks NOx emissions after 1992 (). Prior to that time there is larger uncertainty and probably a bias in the emission estimates that precludes a precise interannual comparison. Trends in ambient precursor data have been reported principally as annual average concentrations or annual averages of maximum daily NO2 concentrations. shows spatially averaged data, representing annual site high-concentration metrics (annual fourth-highest daily peak 8-hr O3 and annual 98th percentile daily maximum 1-hr NO2) averaged across monitoring sites. These high O3 concentrations have not declined as rapidly as the NO2 averages. The 29-site NO2 means are ~10% (range 4–20%) higher than the 98-site NO2 means during a comparable time period (1990–2013), possibly reflecting an earlier emphasis on monitoring NO2 at sites that were more likely to exceed NO2 NAAQS. The 466-site mean O3 concentrations are ~2% (range 0–4%) higher than the 266-site O3 means during a comparable time period (1990–2013).
The observed national-scale O3 declines exhibit statistically significant (p < 0.0001) relationships, with measured NO2 concentrations on an annual basis (). The linear relationship in is relevant to considerations of attainment, but is not necessarily intended to represent the O3 formation and loss processes at finer spatial and temporal resolution. On shorter time scales (i.e., daily and hourly) and finer spatial resolution relevant to individual monitoring sites, O3 concentrations are not typically a linear function of ambient NO2 concentrations. The 1980–2013 national trends are based on fewer monitoring sites than the 1990–2013 trends, and the shorter period yields a y-intercept that is ~5 ppbv lower than the longer period. The relationship shown in is sufficiently robust to the choice of time period and number of trend sites that a sensitivity of national-scale annual-average O3 trends to annual-average ambient NO2 concentrations is evident, with the caveat that the quantified sensitivity is contingent on the aforementioned decreasing anthropogenic VOC emissions. shows that the mean annual fourth-highest daily peak 8-hr O3 concentrations declined approximately linearly with ambient NO2 concentrations, and indicates that the O3 concentration decreases were less than 1:1 proportional to the NO2 declines (i.e., a 1% NO2 decrease yielded less than a 1% O3 decline). If the NOx emission reductions continue downward as projected, then this linear relationship can be extrapolated to low NO2 levels, as supported in the Discussion section later (especially in uncertainties in physicochemical processes). As NO2 concentrations are reduced to zero, we find an intercept of 56 ppbv O3, which approaches background levels cited in the literature. Of course, NO2 has natural sources yielding ambient levels less than 1 ppbv so that reduction in ambient NO2 has to account for a nonzero level.
Figure 3. (a) U.S. national 1980–2013 222-site average of annual fourth-highest daily peak 8-hr O3 vs. 29-site average of the annual 98th percentile concentration of daily 1-hr maximum NO2 concentrations. All regressions are statistically significant (p < 0.0001). (b) National 1990–2013 466-site average of annual fourth-highest daily peak 8-hr O3 vs. 98-site average of the annual 98th percentile concentration of daily 1-hr maximum NO2 concentrations (EPA, Citation2015e, Citation2015f).
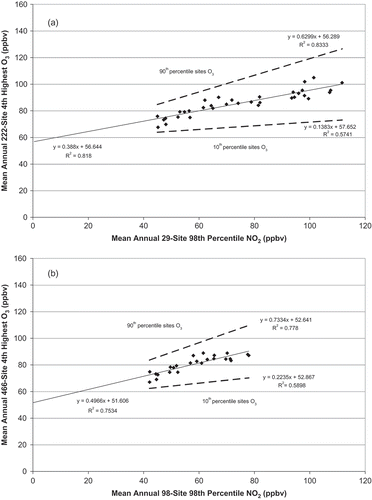
Since O3 concentrations are represented as a function of ambient NO and NO2 concentrations, with sunlight acting on VOCs to generate free radical species, the relationship shown in is plausibly causal. It is also plausibly linear, but not 1:1 proportional, on an annually averaged time scale. However, departures from linearity could occur as ambient NO2 concentrations fall below ~1 ppbv. Mechanistic understanding derived from field and modeling studies, complemented by studies of ambient VOC and NOx trends in specific regions, supports the conclusion that the relationship in depicts causal relationships (Sillman et al., Citation1990; Trainer et al., Citation1993, Citation2000; Kleinman, Citation2000). Modeling studies showed that lowering peak 8-hr O3 concentrations in multiple subregions of central California and the eastern United States to values in the range of 60–70 ppbv would require NOx emission reductions in the range of 80% to over 90% from 1996–1999 emissions (Reynolds et al., Citation2003, Citation2004). In these studies, large (>80% from late 1990s levels) reductions of NOx emissions are required for lowering peak 8-hr O3 concentrations, in part because the effectiveness of the mass reduction of NOx is partially offset by increasing O3 production efficiency at lower ambient concentrations of NOx (Reynolds et al., Citation2004). Numerous other studies have demonstrated that O3 production efficiency (OPE) increases as NOx concentrations decrease (e.g., Lin et al., Citation1988; NARSTO, Citation2000; Pollack et al., Citation2013). Using model-predicted ozone response surfaces, Reynolds et al. (Citation2003, Citation2004) also showed that annual fourth-highest peak 8-hr O3 concentrations would decline approximately linearly with decreasing NOx emissions in a variety of O3 nonattainment areas after region-specific decreases from late-1990s levels of VOC and NOx emissions had occurred.
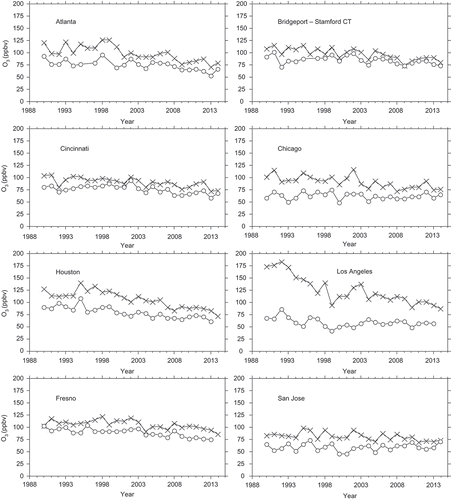
The national-scale data shown in through provide important observational support for photochemical modeling predictions, but lack spatial resolution. Added spatial differentiation is facilitated using the CBSA data. The CBSA trends provide a degree of spatial resolution that is commensurate with understanding O3 attainment (which is typically determined on a multicounty basis). Within individual CBSAs, the trends in the maximum-site annual fourth-highest peak 8-hr O3 concentrations are indistinguishable from linear declines over time, although year-to-year variability precludes definitive characterization of the shapes of the O3 trends (). The minimum-site annual fourth-highest peak 8-hr O3 concentrations generally tracked the maximum-site annual fourth-highest peak 8-hr O3 concentrations within each CBSA, with similar but damped year-to-year variations ().
Figure 4. Trends in annual fourth-highest peak 8-hr O3 at sites having the highest (x) and the lowest (open circles) values each year in eight CBSAs.
Annual trends in the high and low-percentile daily peak 8-hr average O3 concentrations provide insight about O3 response that is not necessarily represented by trends in the current NAAQS—the fourth-highest peak daily 8-hr concentration. As noted earlier, Simon et al. (Citation2015) reported a downward trend in the 95th percentile and the median or mean daily peak 8-hr average O3 concentration, but an increase in the values of the 5th percentile, indicating a compression tendency in the distribution of daily 8-hr O3 maxima (). This also was reported in Lefohn et al. (Citation1998) and in modeling of California and eastern conditions (e.g., Reynolds et al., Citation2003, Citation2004). The principal criterion for the NAAQS is human health risk, linked with a measure of extreme concentrations of O3 historically chosen for the standard in the 1970. Observations suggest that occurrences of the highest peak O3 concentrations have shifted in part outside the historical mid-summer range to spring and fall (e.g., Bloomer et al., Citation2010). Though not addressed here, the response of the full distribution of hourly or peak daily O3 concentrations is relevant, because a form of integral concentration to describe annual cumulative O3 exposure (W126) has been proposed for vegetation exposure (Lefohn, Citation1995). An analogous criterion could evolve as a sustained exposure compared with an 8-hr maximum index, emphasizing a daily midday extreme.
Annual fourth-highest peak 8-hr O3 concentrations exhibited linear decreases with decreasing NO2 concentrations (noting the concurrent declines in VOC emissions), whereas mean O3 concentrations generally did not (). This result suggests that NOx and VOC emission changes have been most effective for reducing the highest O3 concentrations, providing one explanation for the differential O3 trends in . Using different NO2 metrics did not change the O3–NO2 results appreciably. The rates of change in the maximum-site annual fourth-highest peak 8-hr O3 concentrations among CBSAs ranged from 0.6 to 1.6 ppbv O3 per ppbv change in mean annual daily 1-hr peak NO2 concentration (except for Chicago). The maximum site annual fourth-highest peak 8-hr O3 concentration correlated with both the maximum site NO2 and with intersite means of NO2 concentrations, and the results were essentially similar to those shown in except for Chicago (e.g., the annual fourth-highest peak 8-hr O3 vs. intersite mean annual NO2 concentrations r2 values were 0.28, 0.66, and 0.67 for Chicago, Houston, and Los Angeles). Correlations of NO2 concentrations with other high O3 metrics (90th–99th percentiles) were comparable to those shown in . In Atlanta, both high and mean O3 concentrations declined with ambient NO2 concentrations; this was also true for Memphis, TN. The example sites in are typical of other CBSAs within their geographical areas. Compared with Chicago, both Detroit and Milwaukee show more pronounced linear declines of annual maximum site fourth-highest peak 8-hr O3 with NO2 concentrations.
Figure 5. Annual O3 concentration vs NO2 concentration. The O3 metrics are the annual fourth-highest peak 8-hr O3 (x) and the mean peak 8-hr O3 (open circles) concentrations from the maximum O3 site within each CBSA. The NO2 metric is the mean annual daily 1-hr peak NO2 from the site with the maximum mean NO2 concentration within the same CBSA as each O3 site.
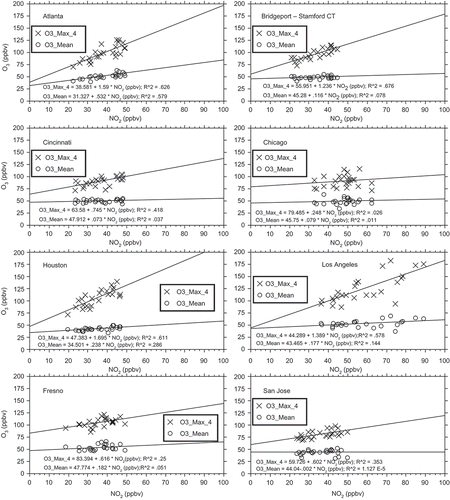
In all cases, the O3 concentration decrease was found to be less than 1:1 proportional to the NO2 decline, yielding statistically significant (p < 0.05) intercepts of 39 to 83 ppbv O3 for the fourth-highest O3 peak daily 8-hr concentration (). The majority of the intercepts were in the range of 50–60 ppbv, consistent with the national average results (). Intercepts for mean O3 versus NO2 concentrations were in the range of 30–50 ppbv (). These results indicate that even at ambient NO2 concentrations approaching zero, a statistical distribution of O3 concentrations will remain due to O3 formation from a so-called “background” of natural or uncontrollable precursors emissions (including NOx, VOC, CO, and CH4), decay chemistry, and transport (including intra- and intercontinental O3 transport).
Summarizing this section, our analysis indicates that the O3–NO2 relationship is statistically strong not only for the national aggregate, but also for most of the local CBSA cases examined. The less than 1:1 proportional relationship between high concentrations of O3 and either NOx emissions or maximum NO2 concentrations is a useful empirical assessment of the effectiveness of NOx controls in terms of the changes in O3 concentration with respect to changes in NOx emissions. The ambient relationships also include the implicit influence of declining NMOC concentrations along with NOx decreases.
Projecting future maximum O3 concentrations
The empirical ambient O3–NO2 relationships provide an opportunity to estimate future O3 levels and to compare them with predictions from photochemical models. (Long-term ambient data have been used by Hogrefe et al. [Citation2011] in retrospective comparisons.) To illustrate such a comparison, we hypothesize that the annual linear O3–NO2 relationship, with a historical range of meteorological conditions, holds at a particular CBSA location as NO2 concentrations decline to near zero. At the same time, NMOC concentrations decline, but do not affect the linear observational relationship. We use modeling results from EPA’s recent predictions supporting its regulatory impact analysis (RIA) for a proposed revision of the O3 NAAQS (EPA, Citation2014a). The regional scale model adopted for the RIA was CAMx, v. 6.1, with appropriate emission projections accounting for known regulatory initiatives as a 2025 baseline, as well as unspecified options to be determined by planners. The emissions projections adopted for the EPA calculations are summarized in Tables S5 and S6.
At a national scale, the RIA modeling predictions underestimate NOx control requirements for revised O3 NAAQS levels compared with the observed 34-year O3–NO2 sensitivity of the 222-trend-site 90th percentile O3 by 4 percentage points (for 70 ppbv) to 18 percentage points (for 60 ppbv) (). The modeling predictions are comparable to the observed 24-year O3–NO2 sensitivity of the 90th percentile O3 among 466 trend sites for 70 ppbv, but underestimate by 9 percentage points for 60 ppbv (). These results suggest that the observed sensitivity of O3 to NO2 is less than the sensitivity that is embedded within the photochemical air quality model. The comparability of observed ambient NO2 trends with NOx emission trends supports extension of inferences from ambient NO2 to NOx emissions. The fit of the national-level O3–NO2 trends is statistically strong (r2 = 0.83 for 34 years or 0.78 for 24 years; ).
Table 3. Comparison of modeling predictions with empirical projections.
The observational analysis did not separate the relative contributions of VOC and NOx reductions to declining O3, so the empirical O3–NO2 sensitivities are contingent on the historical VOC emission decline. EPA’s modeling included VOC impact regions for urban areas with the highest projected 2025-base inventory O3 DVs in each region: New York City, Pittsburgh, and Baltimore in the Northeast; Detroit, Chicago, and Louisville in the Midwest; Houston and Dallas in the Central region; Denver, CO, in the Southwest; and Northern and Southern California. Therefore, the O3–NOx sensitivities for historical observations and for CAMx modeling are both contingent on reductions of anthropogenic VOC emissions. Since future anthropogenic VOC emission reductions are smaller in magnitude than past VOC emission decreases (), the future rates of decline in O3 concentrations with respect to ambient NO2 concentrations could be less than the historical rates but are unlikely to be greater.
The CBSA linear regressions of O3 versus NO2 concentrations are used to predict the ambient NO2 levels at which the annual fourth-highest peak 8-hr O3 at the highest O3 site in each CBSA would reach current (75 ppbv), proposed (70 and 65 ppbv), and alternative (60 ppbv) NAAQS levels. These projections are summarized in and compared with the NOx emission reductions that the EPA modeling studies quantified. The CBSAs listed in were selected because they contain the monitoring sites having the highest projected O3 DVs within each modeling area (EPA, Citation2014a, Tables 3A-7 through 3A-11). Because the RIA modeling focused emission reductions within areas around the sites with highest DVs, the ambient analyses and model predictions in are believed to be as comparable as practical.
The empirical approach illustrates that the observed sensitivity of O3 to NO2 change (with concomitant NMOC change) is less than the sensitivity that is embedded within the photochemical air quality model at one or more CBSAs within each region where the RIA focused emission reductions (Northeast, Midwest, Central, and California). The observations suggest that the modeling predictions underestimate the emission NOx and VOC reductions needed for attaining the present 75 ppbv O3 NAAQS at one or more CBSAs in California and the Central region (), showing larger percentage reductions based on ambient NO2 concentrations than for model-predicted NOx emissions for attaining 75 ppbv O3 in Houston and Dallas–Fort Worth, TX, and Riverside–San Bernardino, Bakersfield, and Fresno, CA. Relative to the ambient data analyses, the modeling predictions also underestimate the emission reductions needed for attaining the possible revised NAAQS of 70, 65, or 60 ppbv O3 in one or more CBSAs in all four emission-control regions (Northeast, Midwest, Central, and California) (). The magnitude of possible underestimation increases as the target O3 concentrations decrease. At 65 ppbv, for example, the maximum listed underestimation is 10 percentage points (Bridgeport–Stamford–Norwood, CT) in the Northeast, 33 percentage points (Cincinnati–Middletown, OH) in the Midwest, 16 percentage points (Dallas–Fort Worth, TX) in the Central region, and 21 percentage points at all listed sites in California.
To check the representativeness of the results listed in , observed sensitivities of O3 to NO2 were determined for all CBSAs having 24 or 25 years of ambient O3 and NO2 measurements. For the 70 ppbv proposed NAAQS, the observations indicate that 25% or more of the 89 CBSAs potentially require greater NO2 reductions than modeling indicates (). The Central, Midwest, Southwest, and Northern California regions have 33% or more of their CBSAs requiring potentially greater NO2 reductions to attain a 70 ppbv O3 NAAQS than the RIA modeling indicates. All regions have 33% or more of their CBSAs requiring greater NO2 reductions to attain a 60 ppbv O3 NAAQS than modeling indicates, and the observations indicate that 52% or more of the 89 CBSAs potentially require greater NO2 reductions for attaining 60 ppbv than modeling indicates.
Table 4. Summary of projected ambient NO2 reductions for meeting four alternative O3 NAAQS levels in 89 CBSAs compared with EPA RIA modeling predictions.
Nearly all (75 of 89) CBSAs showed statistically significant (p < 0.05) O3–NO2 correlations. CBSAs where O3 was less correlated with ambient NO2 concentrations could reflect measurement artifacts, locations where VOC concentrations especially influence O3 formation, or areas where O3 concentrations are dominated by transported O3. CBSAs that are subject to O3 long-range or inter-CBSA transport could experience lower ambient O3 in the future as transported O3 declines.
Given spatially formulated NOx and NMOC emissions, CAMx and other source-based models explicitly address not only parameterized photochemistry, but also meteorological approximations subject to initial and boundary conditions. One could argue that the model predictions provide a more fundamental chemistry-based assessment of NO2 sensitivity than observations. This assertion has not been tested in model comparisons for long-term trends. Hogrefe et al. (Citation2011) have indirectly touched on this issue. In his review, Sillman (Citation1999) has noted the uncertainties embedded in model calculations, and has indicated the importance of comparison with observation-based results characterizing sensitivity regimes. To our knowledge, no tests of the O3–NO2 trend relationships have been addressed in photochemical models to evaluate the linearity hypothesis. Our observational hypothesis implicitly factors photochemistry, meteorological variation, boundary conditions, and short-range transport into the results by relating the maximum O3 concentrations among sites within a CBSA (i.e., typically a downwind location) to the maximum observed NO2 concentrations among sites within the same CBSA including urban core areas). Potential limitations and uncertainties in the empirical relationships are discussed in detail in the next section.
In summary, the results illustrate the capability of using empirical trends from ambient data as a means of checking the predictions of photochemical modeling, given key data on precursor emissions and meteorology over a span of time. The results suggest that the model application (CAMx, v. 6.1) could have greater O3 sensitivity to NOx reductions than the actual ambient data.
Discussion
NMOC data limitations
As noted, the O3–NO2 sensitivities discussed here are contingent on the concomitant effects of a historical VOC emission decline. Potentially different O3–NO2 sensitivities might have resulted if anthropogenic VOC emissions had been constant over time. Tracing the reduction in ambient NMOC with VOC emissions is problematic given the limited data from the PAMS network, and the known large uncertainties in the reported NMOC concentrations. However, insight into the regional distribution of NMOC and trends can be obtained from the PAMS sites listed in Table S1. The areas nominally covered by PAMS include California, the southern U.S. border, the southern Great Lakes, and the greater Northeast. The coverage is generally daily observations in summer, as hourly or multihourly samples, and 24-hr average canister samples collected once every third or sixth day throughout the year. The sampling record is from the early 1990s to the present. The sites include some of EPA’s designated climate regions of the United States, but not all of them. The PAMS data are known to be variable in quality and reliability even though they are nominally obtained with the same instrumentation and the same laboratory, quality control, and assurance practices (e.g., Parrish and Fehsenfeld, Citation2000). Direct relationships between VOC emissions and ambient NMOC concentrations have been demonstrated for examples such as southern California (Pollack et al., Citation2013) and the Southeast (Hidy et al., Citation2014). Examples of such relationships are shown in the regression results of Table S4 for the Southeast. Here, O3 trends for Atlanta, GA, and nonurban sites show significant relations (p < 0.05) with motor vehicle VOC emissions, but a significant relation is not observed in Birmingham, AL. The rate of change in O3 per mass of VOC reductions tends to exceed that of NOx in the table.
The difficulty in applying PAMS data by region can be illustrated using trends estimated from annual averages of PAMS indicator species.
We have selected from the reported 56 PAMS species those compounds that are believed to be analytically stable to sampling and analytical errors and are representative of different major anthropogenic and natural categories. Those shown in Figure S4 are (a) sum of C2–C4 (C234) hydrocarbons representing natural gas indicators, petroleum processing, and mobile or gasoline sources, (b) sum of pentanes (C5), mainly mobile sources, (c) sum of benzene, toluene, ethyl benzene, and xylenes (BTEX) aromatic compounds, mainly gasoline and to a much lesser extent industrial sources, and (d) isoprene, a natural emission indicator. The NMOC data indicate generally a downward trend in BTEX and C2–C4 hydrocarbons (except for the Southeast and South; the South region has large petroleum and petrochemical industries). The pentanes show weak or no decline except for the Southeast, where mean pentane concentrations erratically increase over the period for an unknown reason. Mean isoprene tends to be less than ~2 ppbv in all regions except the Southeast and is approximately constant from 2000 to 2012; the measurements suggest that mean isoprene concentrations tend to increase in the Southeast and southern California (see also Hidy et al., Citation2014; Pollack et al., Citation2013). The elevated levels of isoprene in the Southeast indicate the known strong potential influence of isoprene and associated natural VOCs such as the terpenes as major factors in O3 production in the Southeast compared with the rest of the United States. We conclude from inspection of the PAMS data that the indicators selected for anthropogenic NMOC species tend to show a decline consistent with the NEI nationally and by region. However, this decline is much less “convincing” than the ambient NO2 trends.
Taking into account the limitations of the NMOC data, insight into the strength of the NO2–O3 relationship relative to NMOC can be obtained from trends. This is facilitated by the fact that most of the VOC emissions in urban areas derive from motor vehicles (a potential exception is Houston, TX [e.g., Couzo et al., Citation2013]). Toluene is frequently used as an indicator for motor vehicle NMOC (e.g., Pollack et al., Citation2013); we extracted and annually averaged toluene from the PAMS data set. We conducted univariate O3–NO2 and O3–toluene concentration regressions, and multivariate regressions for 26 example CBSA cases (augmenting the original set of 24 CBSAs with examples having more complete toluene data). The univariate regressions for O3 versus NO2 concentrations were statistically significant (p < 0.05) for 22 of the 26 CBSAs. The univariate regressions of O3 versus toluene were statistically significant for 17 CBSAs. The multivariate regressions of O3 versus NO2 and toluene concentrations were statistically significant (p < 0.05) for NO2 for 16 and toluene for 5 CBSAs. For the univariate regression the fit to the data was better for NO2 than for toluene for 23 of the examples.
The regressions are illustrated in Figure S5 for Los Angeles, Chicago, Bridgeport–Stamford, CT (New York City-related), and Atlanta, which are believed to be strongly influenced by motor vehicle VOC emissions. Los Angeles is known to be a VOC-sensitive regime and Chicago is suspected to have the same condition. Atlanta through 2007 was largely VOC sensitive, but has moved toward NOx sensitivity since the city is embedded within a regional NOx sensitive environment. The southern New York–Connecticut area is likely to be moving toward NOx sensitivity, especially with a large O3 transport component influencing this area. The selection in Figure S5 illustrates the overall regression results, which show stronger correlations of high O3 with mean NO2 than with mean toluene concentrations, except for Chicago and southern California. The regressions and graphical analyses suggest that future VOC reductions from motor vehicles will have more limited influence on lowering peak O3 concentrations toward NAAQS goals than have past VOC reductions: Because the mean ambient toluene concentrations declined to dramatically low levels between 1990 and 2013, the magnitude of future reductions of ambient concentrations will necessarily be much smaller.
We infer from this analysis that, in aggregate, ambient NO2 concentrations are a stronger predictor of O3 response than are ambient concentrations of toluene, a representative of a large contributor to NMOC. While the NO2–O3 concentration relationship is present as a chemical driver in all regimes (see also “Uncertainties,” discussed next), the response of the highest O3 concentrations to NO2 is expected to be strongest in a NOx-sensitive regime (e.g., Figure S6, higher ratios of VOC/NOx). Historical emission reductions have tended to increase the emissions ratio of VOC/NOx, in principle shifting O3 response toward NOx limitation on more days and over larger geographical areas. Future VOC emission reductions are therefore unlikely to shift the observed O3–NO2 regressions toward a detectable nonlinearity.
Uncertainties deriving from emissions
Projecting changes in O3 in response to precursor reductions requires interpretation, accounting for uncertainties in precursor emissions estimates, and addressing details in precursor reductions in place or planned. Emission uncertainties depend on the contributors to estimation magnitude and error reflected in the reported inventories, while interpretation depends on photochemical relationships established from theory and experiment. Guidance for continued reduction in O3 concentrations focuses on NOx reduction in keeping with the direction foreseen from theory and ambient data interpretation (e.g., Sillman et al., Citation1990; National Research Council [NRC], 1991; Trainer et al., Citation2000; NARSTO, Citation2000).
Projections for conditions based on the relationship between O3 concentrations and NO2 depend on precursor reductions extending in a similar way into the future. These in turn will depend on reliability of NOx and anthropogenic VOC emissions and their projections, as well as influence of changes in chemical response and approach to a background O3 and precursor level. Within the limitations of emission estimates and air quality modeling, projections in O3 trends relative to precursor emissions tend to level out through 2025. This is found in the national projections, and is exemplified in the California projections shown in Figure S3. In the California examples, emission rates of both O3 precursors tend to level off after 2015. Similar national estimates accounting for reductions in place for the next decade show this leveling in emissions as well, as planned programs achieve their goals. These reported projections imply a difficulty in maintaining the linear rate of decline of O3 with precursors as seen in the historical observations.
Uncertainty in reduction of precursor emissions projected into the future introduces uncertainties in the future rate of O3 decline. In the case of NOx, the uncertainties are likely to be associated with two major source categories: combustion sources, including motor vehicles, and electricity generation. Anthropogenic VOC is identified principally with motor vehicles, and to a lesser extent petroleum processing and natural gas production. For motor vehicles, estimates and projections from the NEI depend on the reliability of the MOVES and nonroad source (NONROAD) emission models; assumptions about emission controls (e.g., McDonald et al., Citation2013), fleet makeup, and annual turnover are essential to projections of these emissions over the next decade. Projections of emissions from the vehicle sector indicate precursor reductions, but limited shifts in precursor ratios are expected over the next decade. Fossil fuel combustion emissions in electric power generation depend on projected shifts in generation technology away from fossil fuels, particularly coal; these changes will affect NOx reductions, but are likely to differ marginally from current planned sector changes in the next decade.
We assume that the local ambient NO2 concentrations are indicative of regional emission changes. Assumptions for projecting future emission controls depend on the extent of interstate transport, including southern Canadian emissions towards the Northeast, Mexican emissions affecting the southern United States–northern Mexico border region, and Asian pollution in air entering the West Coast. We assume that these spatial relationships for emissions and meteorology remain similar or the same over the next decade. As noted, regional changes in emission patterns such as the NOx reductions from the 2002 NOx SIP call have not appeared to change the O3 reduction rate in affected eastern locations.
Uncertainties in physicochemical processes
Factors embedded in O3 photochemistry also affect the O3 decline rate. A known relationship exists between O3 and NOz (or NOy), whose ratio is a measure of ozone production efficiency (OPE). In general, as NOx emissions are reduced, the OPE tends to increase (e.g., Lin et al., Citation1988; Kleinman, Citation2000; NARSTO, Citation2000; Pollack et al., Citation2013), which can account for O3 reductions less than proportional. In rural conditions, the OPE is higher than in urban conditions. Increases in OPE have been seen not only in California but also in most other locations, including the Southeast (e.g., Blanchard et al., Citation2014a). While the rural OPE tends to increase, theory also indicates that below a certain NO2 or NOy concentration the efficiency declines to zero (e.g., Lin et al, Citation1988; Thornton et al., Citation2002). The reason for this change is related to the fact that at very low NO2 concentrations O3 production goes toward zero. The regime where O3 production decreases is believed to be at <1 ppbv NO2. Exploration of CBSA data in a relatively low range of NO2 concentrations with mean 1-hr maximum concentrations of 10 ppbv or less indicates no departure in linearity of the O3–NO2 relationships. Examples are shown in Figure S7. Since our projected 1-hr maximum NO2 levels of interest are well above 1 ppbv, especially during high O3 production cycles, nonlinearity over a range of NO2 conditions appears to absent except perhaps in remote locations irrelevant to O3 management.
It is problematic to expect a more aggressive annual reduction rate in (high) O3 concentrations beyond the current precursor proportionality since aggressive measures for NOx and VOC reduction are in place and have not produced an accelerated O3 concentration reduction beyond that prior to the mid-2000s. At the same time, receptor-based modeling of the Atlanta area using a generalized additive model (GAM) (Blanchard et al., Citation2014a) indicates O3 sensitivity to NOy but not to NMOC, at least in the Southeast, covered for example, by the Southern Aerosol Research and Characterization (SEARCH) project. The Downey et al. (Citation2015) calculations for Sacramento, CA, Los Angeles, CA, Philadelphia, PA, and St. Louis, MO, using the Community Model for Air Quality (CMAQ) also suggest regional O3 dependence on NOx reductions relative to NMOC.
The projected emission reductions at the national and some state levels through 2025 appear to be at best similar to those achieved over the past two decades. A continued precursor reduction at the same historic linear rate as seen since 1996 could roughly result in about 10 ppbv maximum O3 change over the next ten years. Reduction in annual fourth-highest daily peak 8-hr average O3 concentration over 3 years as listed in Table S2 would suggest that the cited areas could realize O3 concentrations of ~60–70 ppbv. Accelerated O3 reduction with the NOx–NMOC concentration reductions could occur but would depend on nonlinear interactions of NOx and NMOC with NMOC/NO2 ratios that are absent from the historical data record. A less than 1:1 proportional decrease of O3 concentrations with NO2 exemplified in the SoCAB potentially can be viewed as a nonlinear response to reduction in precursors. This effect can be seen qualitatively for the SoCAB using the well-known “Haagen–Smit” or “EKMA” diagram. An example in Figure S6 was calculated from a photochemical model applied to 1999–2000 for the Los Angeles airshed (Fujita et al., Citation2003). The diagram relates VOC (NMOC) and NOx concentration to O3 in the form of O3 isopleths. The relationships in Figure S6 show the complexity in O3 response to NOx and VOC changes. The regime in the early 2000s for the SoCAB is VOC sensitive, but also responds weakly to NOx change. Hypothetically, if an NOx-sensitive regime to the lower right in Figure S6 exists, one could obtain qualitatively a near-linear O3 reduction with NO2 reduction over a limited range of O3 concentrations. Generally, the projections for NOx and NMOC reductions appear to move conditions toward increasing NOx sensitivity in most regions of the United States. If extreme O3 concentrations prevail as NOx or NMOC sensitive, one deduces that a near-linear relationship between precursor and the highest O3 concentrations may exist in the observations. If precursor concentrations reach an NOx-sensitive regime, say for ambient (molar) NMOC/NO2 > 10, NOx reductions become the main control pathway for O3 reduction.
Hypothetically, measurement-based projections of O3 reductions from reductions in precursor concentrations will depend on maintaining approximately similar local (or regional) conditions in the VOC/NOx ratio. The U.S. trend in mass emission ratio since 2002 is ~0.8 increase in VOC/NOx, with regional levels 0.4–1.1 (lower values in Central and West North Central Regions; e.g., Blanchard and Hidy, Citation2013, emissions from Figure S2) through 2012. The national emission ratio has remained steady for 2002–2012. In the EPA regions, the industrial East, Central, and Pacific Coast regions have remained steady, while the East and West Central regions and South to Southwest regions have increased. This suggests a move toward more NOx sensitive conditions in these latter regions (Figure S2).
Significance of background
As NOx and NMOC concentrations are reduced, ambient O3 concentrations will tend to asymptotically level to a background concentration, nominally toward the zero–NOx “intercept” of the O3–NOx response relationship. As discussed earlier, the regression O3 concentration intercepts tend to be higher than background levels reported in the literature, thus suggesting a potential “ambiguous” response of the highest peak O3 concentrations if ambient NO2 concentrations fall below ~1 ppbv.
Background concentrations of ground-level O3 have been investigated for some time, especially in terms of its rising levels across North America (e.g., Vingarzan, Citation2004; Oltmans et al., Citation2006, Citation2013; Fiore et al., Citation2014). EPA (Citation2012) has addressed a background level in the United States using modeling estimates supported by work of Emery et al. (Citation2012). More recently, Lefohn et al. (Citation2014) have estimated background O3 levels in several U.S. cities. Lefohn et al. (Citation2014) note that the U.S. continental background should be defined with two components, a global component and a North American continental component that includes the chemical interactions with anthropogenic and natural precursor emissions over multispatial scales. Both seasonally and spatially estimated O3 background levels vary. Their definition of background separates a global background from a precursor emission-influenced background (EIB) that involves interactions of background O3 with continental emissions of precursors and O3. The presence of regional or continental background concentrations will tend to influence the level of O3 reduction to be achieved controlling precursor emissions. Their estimates of the fraction of EIB of total urban O3 concentrations based on modeling are listed for spring and summer in Table S7. This part of background may change over the United States with reductions in NOx and VOC emissions. The urban O3 concentrations remain high compared with nominal background estimates of 20–50 ppbv; however, rural O3 levels are believed to be closer to background estimates, especially in the West. The consequence of approaching an “unmanageable” background could realize an overall O3 precursor proportionality trend of even less than 1:1 proportionality.
The many studies of O3 background have settled on variable levels depending on the impact of long-range transport, especially in the West—continental and intercontinental transport of O3 and its precursors (e.g., Oltmans et al., Citation2008; Parrish et al., Citation2009). There also is stratospheric O3 intrusion, especially in the spring, that influences O3 background (e.g., Lefohn et al., Citation2014). Modeling estimates of background have been discussed in EPA’s (Citation2014a) recent RIA.
Virtually all of the literature discussion on background has focused on O3. However, oxidant chemistry at background level needs to include the implications of NOy and NMOC as well. Background levels for NOy and NMOC and their relevance to O3 production have not been addressed extensively in the literature. NOy background derives from sources such as soil respiration and lightning, as well as an intra- and intercontinental residual (e.g., Guenther et al., Citation2000; Emmons et al., Citation1997; Bradshaw et al., Citation2000). For the marine background, NOx levels are at the 10–100 pptv level. For the North American continent, NOx background appears to range between 0.1 ppbv and <1 ppbv, with the latter derived from measurements in the eastern United States where transport of pollution is a factor (e.g., Ridley, Citation1991). NMOC background relates to natural emissions, principally from biota (vegetation). NMOC from vegetation varies widely with temperature. For summer conditions, isoprene, for example, in the Southeast is approximately >10 ppbv during the day, but much less in other regions (e.g., Figure S4). Isoprene is typically perhaps 10% of total vegetation emissions. Under hypothetical natural conditions one expects that conditions for O3 production involve an extreme of NMOC/NOx ratio, or a high degree of NOx sensitivity.
Continental-scale modeling has elucidated the important role of CO and CH4 in producing tropospheric O3 as part of the “slow” photochemical cycle relative to NMOC and NOx that form the “fast cycle” (e.g., Seinfeld and Pandis, Citation1998). The background concentrations of precursor will maintain O3 concentrations at some level and will tend to support the observed O3 background levels.
Conclusions
This study has revealed certain results about combined trends of O3 and its precursors. These are:
While long-term aggregated U.S. national and regional O3 precursor data are limited in spatial coverage, examples indicate that NOx and VOC precursor emissions and ambient concentrations track one another since the 1990s.
Ozone concentration decreases after the 1990s are associated with both NOx and NMOC concentration reductions, even though NOx reductions have been larger than NMOC reductions. Decreases are most pronounced at high O3 concentrations, and weaker or nonexistent at median and lower concentrations.
Observations of O3, NO2, and NMOC concentration changes since the mid-1990s, combined with estimated emission reductions, indicate that the effectiveness of O3 controls in U.S. urban–suburban locations is less than 1:1 proportional to precursor emissions and associated ambient concentrations.
Trend measurements focus on high O3 concentrations in the 98th percentile range at or near NAAQS nonattainment areas. They do not show by decade different rates of change in O3 with declining NO2 between the 1980s and 2000s. Overall, the 1980 to post-2000 data fit a linear trend as well as other assumed forms. The consistently sustained decrease indicates that the long-term O3 decline rate has not accelerated after 2002 with implementation of EPA’s NOx SIP call and other mandated reductions of NOx and VOCs.
Projected rates of O3 decline will not exceed past rates over the next decade, based on extrapolation of the NO2–O3 relationships. If the current linearity in O3 response to NO2 continues with concomitant VOC reductions, the prospects are limited for achieving proposed revised NAAQS targets of 65 ppbv or less for the annual fourth-highest daily 8-hr peak O3 concentrations. Measurements do not indicate increasing annual reduction rates in (high) O3 concentrations beyond the current precursor proportionality, since aggressive measures for NOx and VOC reduction are in place and have not produced accelerated O3 concentration reduction beyond that prior to the mid-2000s.
A comparison of projections from CBSA empirical O3–NO2 relationships with results from the CAMx photochemical model indicates how ambient data relationships can check the results from US regional modeling for sensitivity of changes to NOx emissions. An example for U.S. national projections to 2025 shows that version 6.1 of the model tends to be more sensitive to precursor emission reductions than supported by O3–NO2 observations to date.
Acknowledgments
The authors thank John Jansen for his review and insightful comments on the paper.
Funding
This study was supported by Southern Company.
Supplemental Data
Supplemental data for this paper can be accessed at the publisher’s website.
Supplemental Material
Download Zip (2 MB)Additional information
Funding
Notes on contributors
George M. Hidy
George M. Hidy is a principal of Envair/Aerochem.
Charles L. Blanchard
Charles L. Blanchard is a principal of Envair.
References
- Aleksic, N., J.-Y. Ku, and L. Sedefian. 2013. Effects of the NOx SIP Call program on ozone levels in New York. J. Air Waste Manage. Assoc. 63:1335–43. doi:10.1080/10962247.2013.824392
- Bergin, M., J.-S. Shih, A. Krupnik, J. Boylan, J. Wilkinson, and A. Russell. 2007. Regional Air quality and interstate impacts of NOx and SO2 emissions on ozone and fine particulate matter in the Eastern United States. Environ. Sci. Technol. 41:4677–89. doi:10.1021/es062302s
- Blanchard, C., and G. Hidy. 2013. How does ozone respond to VOC and NOx emissions? Unpublished report prepared for Hunton and Williams. In Comments of the Utility Air Regulatory Group on EPAs proposed rule to implement the 2008 ozone NAAQS: State Implementation Plan requirements, ed. L. Langworthy and T. Szymanski. Docket ID No. EPA-HQ-QAR-2010-0885. Washington, DC: Hunton and Williams, LLC.
- Blanchard, C., S. Tanenbaum, G. Hidy, R. Rasmussen, and R. Watkins. 2010a. NMOC, ozone and organic aerosol in the southeastern states, 1999–2007. 1. Spatial and temporal variations of NMOC concentrations and composition in Atlanta, Georgia. Atmos. Environ. 44:4827–39. doi:10.1016/j.atmosenv.2010.08.036
- Blanchard, C., S. Tanenbaum, and G. Hidy. 2010b. NMOC, ozone and organic aerosol in the southeastern states. 1999–2007: 2. Ozone trends and sensitivity to NMOC emissions in Atlanta, Georgia. Atmos. Environ. 44:4840–49. doi:10.1016/j.atmosenv.2010.07.030
- Blanchard, C., S. Tanenbaum, and G. Hidy. 2014a. Ozone in the southeastern United States: An observation-based model using measurements from the SEARCH network. Atmos. Environ. 48:192–200. doi:10.1016/j.atmosenv.2014.02.006
- Blanchard, C., S. Tanenbaum, and G. Hidy. 2014b. Source attribution of air pollutant concentrations and trends in the Southeastern Aerosol Reserarch and Characterization (SEARCH) network. Environ. Sci. Technol. 47:13,536–45.
- Bloomer, B., K. Vinnikov, and R. Dickenson. 2010. Change in seasonal and diurnal cycles of ozone and temperature in the eastern U.S. Atmos. Environ. 44:2543–51. doi:10.1016/j.atmosenv.2010.04.031
- Bradshaw, J., R. Newell, E. Sandholm, and S. Liu. 2000. Observed distributions of nitrogen oxides in the remote free troposphere from the NASA Global Tropospheric Experiment Programs. Rev. Geophys. 38:61–116. doi:10.1029/1999RG900015
- Bureau of Air Quality Analysis and Surveillance. 2007. Trends in measured 1-h ozone concentrations over the OTR modeling domain. Albany, NY: New York Department of Environmental Conservation.
- Butler, T., M. Vermeylen, G. Rury, G. Likens, D. Lee, G. Bowker, and L. McCluney. 2011. Response of ozone and nitrate to stationary source NOx emission reductions in the eastern USA. Atmos. Environ. 45:1084–94. doi:10.1016/jj.atmosenv.2007.04.061
- California Air Resources Board (CARB). 2014a. Air quality data. www.arb.ca.gov/aqd/almanac/almanac/htm ( accessed January, 2015) .
- California Air Resources Board (CARB). 2014b. Air quality trend summaries. http://www.arb.ca.gov/adam/trends/trends1.php ( accessed January 2015).
- California Air Resources Board (CARB). 2015a. The California almanac of emissions and air quality. http://www.arb.ca.gov/aqd/almanac/almanac.htm (accessed January 2015).
- California Air Resources Board (CARB). 2015b. CEPAM: 2013 Almanac—Standard emissions tool. Emission projections by summary category. http://www.arb.ca.gov/app/emsinv/fcemssumcat2013.php (accessed January 2015).
- California Air Resources Board (CARB). 2015c. Air quality trend summaries http://www.arb.ca.gov/adam/trends/trends1.php (accessed January 2015).
- Camalier, L., W. Cox, and P. Dolwick. 2007. The effects of meteorology and their use in assessing ozone trends. Atmos. Environ. 41:7127–37. doi:10.1016/j.atmosenv.2007.04.061
- Chan, E. 2009. Regional ground-level ozone trends in the context of meteorological influences across Canada and the eastern United States from 1997–2006. J. Geophys. Res. Atmos. 114:D05301. doi:10.1029/2008JD010090
- Connecticut Department of Energy and Environmental Protection (Connecticut DEEP). 2009. State 8 hour ozone implementation plan, 2009. Sect. 3.0 Ozone air quality levels in Connecticut and recent trends. http:// www.ct.gov/deep/cwp/view.asp?a=26848&q=385886AdeepNav_GID=1619 ( accessed February 2014).
- Cooper, O., R.-S. Gao, D. Tarasick, T. Leblanc, and C. Sweeney. 2012. Long-term ozone levels at rural ozone monitoring sites across the United States, 1990–2010. J. Geophys. Res. Atmos. 117: D22307.
- Cooper. O., D. Parrish, J., Ziemke, N. Balashov, M. Cupeiro, I. Galbally, S. Gilge, I. Horowitz, N. Jensen, J. Larmarque, N. Naik, S. Oltmans, J. Schwab, D. Shindell, A. Thompson, V. Thouret, Y. Wang, and R. Zhinden. 2014. Global distribution and trends of tropospheric ozone: An observation-based review. Elementa 2:1–28. doi:10.12952/journalelementa.000029
- Couzo, E., H. Jeffries, and W. Vizuete. 2013. Houston’s rapid ozone increases: preconditions and geographic origins. Environ. Chem. 10:260–88. doi:10.1071/EN13040
- Department of Environmental Conservation, New York State (DEC NYS). 2013. Air quality trends. Albany, NY: New York Department of Environmental Conservation. www.dec.ny.gov/chemical/54359.html ( accessed July 2013) .
- Demerjian, K. 2000. A review of national monitoring networks in North America. Atmos. Environ. 34:1861–85. doi:10.1016/S1352-2310(99)00452-5
- Downey, N., C. Emery, J. Jung, S. Sakulyanontvittya, L. Hebert, D. Blewitt, and G. Yarwood. 2015. Emission reductions and urban ozone responses under more stringent US standards. Atmos. Environ. 101:209–16. doi:10.1016/j.atmosenv.2014.11.018
- Emery, C., J. Jung, N. Downey, J. Johnson, M. Jimenez, G. Yarwood, and R. Morris. 2012. Regional and global modeling estimates of policy relevant background ozone over the United States. Atmos. Environ. 47:206–17. doi:10.1016/j.atmosenv.2011.11.012
- Emmons, L., M. Carroll, D. Hauglustine, G. Brasseu, C. Atherton, J. Penner, S. Sillman, H. Levy II, F. Rohrer, W. Wauben, P. Van Velthoven, Y. Wang, D. Jacob, P. Bakwin, R. Dickerson, B. Dodderidge, C. Gerbig, R. Honrath, G. Hubler, D. Jaffe, Y. Kondo, J. Munger, A. Torres, and A. Volz-Thomas. 1997. Climatologies of NOx and NOy: A comparison of data and models. Atmos. Environ. 31:1851–904. doi:10.1016/S1352-2310(96)00334-2
- Environmental Protection Agency. 1998. Findings of significant contributions and rule making for certain states in the Ozone Transport Assessment Group Region for purposes of reducing regional transport of ozone (US). Fed. Reg. 63:57356–38.
- Environmental Protection Agency. 2005. Ozone assessment through 2004. Evaluating NOx and VOC control programs in the eastern United States, focus on the NOx Budget Trading Program, 2004. EPA-454-K-05-001. Research Triangle Park, NC: Office of Research and Development.
- Environmental Protection Agency. 2012. Regional and seasonal analysis of North American background ozone estimated from two studies. http://www.epa.gov/ttn.naaqs/standards/ozone/data/20120814BackgroundOzone.pdf ( accessed January 2015)
- Environmental Protection Agency. 2013a. 2011 National Emissions Inventory: Data and documentation. http://www.epa.gov/ttn/chief/net/2011inv.html (accessed June 2013).
- Environmental Protection Agency. 2013b. Trends in ozone adjusted for weather conditions. http://www.epa.gov/airtrends/weather.html (accessed October 6, 2013).
- Environmental Protection Agency. 2014a. Regulatory impact analysis for the proposed revisions of the National Ambient Air Quality Standard for ozone. http.www.epa.gov/ttn/naaqs/standards/ozone/a_o3_ria.html ( accessed January, 2015).
- Environmental Protection Agency. 2014b. PAMS network and sites. http://www.epa.govttnamti1/pamssites.html (accessed December 14, 2014).
- Environmental Protection Agency. 2015a. National Ambient Air Quality Standards (NAAQS). http://www.epa.gov/air/criteria.html (accessed January 2015).
- Environmental Protection Agency. 2015b. Technical support document (TSD): Preparation of emissions inventories for the version 6.1, 2011 Emissions Modeling Platform. http://www.epa.gov/ttn/chief/emch/2011v6/2011v6.1_2018_2025_base_EmisMod_TSD_nov2014_v6.pdf (accessed January 2015).
- Environmental Protection Agency. 2015c. ftp://ftp.epa.gov/EmisInventory/2011v6/ozone_naaqs/reports/ and ftp://ftp.epa.gov/EmisInventory/2011v6/v1platform/reports/2018_emissions/ ( accessed January 2015).
- Environmental Protection Agency. 2015d. National Emissions Inventory (NEI) air pollutant emissions trends data. http://www.epa.gov/ttn/chief/trends/index.html (accessed January 2015).
- Environmental Protection Agency. 2015e. National trends in nitrogen dioxide levels. http://www.epa.gov/airtrends/nitrogen.html (accessed January 2015).
- Environmental Protection Agency. 2015f. National trends in ozone levels. http://www.epa.gov/airtrends/ozone.html (accessed January 2015).
- Fiore, A., D. Jacob, J. Logan, and J. Yin. 1998. Long-term trends in ground level ozone over the contiguous United States, 1980–1995. J. Geophys. Res. 103:1471–80. doi:10.1029/97JD03036
- Fiore, A. J. Obreman, M.-Y. Lin, L. Zhang, O. Clifton, D. Jacob, V. Naik, L. Horowitz, J. Pinto, and G. Milly. 2014. Estimating North American background ozone in U.S. surface air with two independent global models. Variability, uncertainties, and recommendations. Atmos. Environ. 96:284–300. doi:10.1016/j.atmosenv.2014.07.045
- Fujita, E., W. Stockwell, D. Campbell, R. Keisslar, and D. Lawson. 2003. Evolution of the magnitude and spatial extent of the weekend ozone effect in California’s South Coast Air Basin, 1981–2000. J. Air Waste Manage. Assoc. 53:802–15. doi:10.1080/10473289.2003.10466225
- Fujita, E.M., D.E. Campbell, B. Zielinska, J.C. Chow, C.E. Lindhjem, A. DenBleyker, G.A. Bishop, B.G. Schuchmann, D.H. Stedman, and D. R. Lawson. 2012. Comparison of the MOVES2010a, MOBILE6.2 and EMFAC2007 mobile source emissions models with on-road traffic tunnel and remote sensing measurements. J. Air Waste Manage. Assoc. 62:1134–49. doi:10.1080/10962247.2012.699016
- Gego, E., P. Porter, A. Gilliland, and S.T. Rao. 2007. Observation-based assessment of the impact of nitrogen oxides emissions reductions on ozone air quality over the eastern United States. J. Appl. Meteorol. Climatol. 46:994–1008. doi:10.1175/JAM2523.1
- Gilliland, A., C. Hogrefe, R. Pinder, R. Godowitch, K. Foley, and S.T. Rao. 2008. Dynamic evaluation of regional air quality models: Assessing changes in O3 stemming from changes in emissions and meteorology. Atmos. Environ. 42:5110–23. doi:10.1016/j.atmosenv.2008.02.018
- Godowitch, J., A. Gilliland, R. Draxler, and S.T. Rao. 2008. Modeling assessment of point source NOx emission reductions on ozone levels in the eastern United States. Atmos. Environ. 42:87–100. doi:10.1016/j.atmosenv.2007.09.032
- Guenther, A., C. Geron, T. Pierce, B. Lamb, P. Harley, and R. Fall. 2000. Natural emissions of non-methane volatile organic compounds, carbon monoxide, and oxides of nitrogen from North America. Atmos. Environ. 34:2205–30. doi:10.1016/S1352-2310(99)00465-3
- Harley, R., S. Giddings, and L. Marr. 2004. Decadal trends in air pollutant emissions from motor vehicles in Central California. Report 00-14CCOS, Figure 4. Sacramento, CA: California Air Resources Board. www.arb.ca.gov/airways/cos/docs/II4_0014_May04_fr.pdf (accessed March 2015).
- He, H., J. Stehr, J. Hains, D. Krask, B. Doddridge, K. Vinnikov, T. Canty, K. Hosley, R. Salawitch, H. Worden, and R. Dickerson. 2013. Trends in emissions and concentrations of air pollutants in the lower troposphere in the Baltimore/Washington airshed from 1997–2011. Atmos. Chem. Phys. 13:7859–74. doi:10.5194/acp-13-7859-2013
- Hidy, G., J. Brooks, K. Demerjian, L. Molina, W. Pennell, and R. Scheffe. 2011. Technical challenges of multipollutant air quality management. New York, NY: Springer.
- Hidy, G., C. Blanchard, K. Baumann, E. Edgerton, S. Tanenbaum, S. Shaw, E. Knipping, I. Tombach, J. Jansen, and J. Walters. 2014. Chemical climatology of the southeastern United States, 1999-2013. Atmos. Chem. Phys. 14:11893–914. doi:10.5194/acp-14-11893-2014
- Hogrefe, C., W. Hao, S. Zalewsky, J.-Y. Ku, B. Lynn, C. Rozenzweig, M. Schultz, S. Rast, M. Newchurch, L. Wang, P. Kinney, and G. Sisila. 2011. An analysis of long-term regional-scale ozone simulations over the Northeastern United States: Variability and trends. Atmos. Chem. Phys. 11:567–82. doi:10.5194/acp-11-567-2011
- Jing, P., Z. Lu, J. Xing, D. Streets, Q. Tan, T. O’Brien, and J. Kamberos. 2014. Response of the summertime ground-level ozone trend in the Chicago area to emission controls and temperature changes, 2005–2013. Atmos. Environ. 99:630–40. doi:10.1016/j.atmosenv.2014.10.035
- Klasinc, L., N. Kezele, M. Pompe, and S. McGlynn. 2008. Trends, distribution and frequency analysis of ozone data from three monitoring stations in Baton Rouge, Louisiana for the years 1995 to 2005. Croatica Chemica Acta 81:311–18.
- Kleinman, L. 2000. Ozone process insights from field experiments—Part II. Observation-based analysis for ozone production. Atmos. Environ. 34:2023–34. doi:10.1016/S1352-2310(99)00457-4
- Koerber, M. 2010. Review of Midwest Air Quality. Lake Michigan Air Directors Consortium (LADCO). http://www.Ladco.org/reports/workshops/2010/October_27_2010/Koerber.pdf ( accessed January 2015).
- Lefohn, A. 1995. How the W126 ozone exposure index was developed. www.asl-associates/w126.htm ( accessed January 2015).
- LeFohn, A., J. Shadwick, and S. Ziman. 1998. The difficult challenge of attaining EPA’s new ozone standard. Environ. Sci. Technol. 32:276A–82A. doi:10.1021/es983569x
- Lefohn, A., D. Shadwick, and S. Oltmans. 2010. Characterizing changes in surface ozone levels in metropolitan and rural areas in the United States for 1980–2008 and 1994–2008. Atmos. Environ. 44:5199–210. doi:10.1016/j.atmosenv.2010.08.049
- LeFohn, A., C. Emery, D. Shadwick, H. Wernli, J. Jung, and S. Oltmans. 2014 Estimates of background surface ozone concentrations in the United States based on model-derived source apportionment. Atmos. Environ. 84:275–88. doi:10.1016/j.atmosenv.2013.11.033
- Lin, X., M. Trainer, and S. C. Liu. 1988. On the nonlinearity of the tropospheric ozone production. J. Geophys. Res. Atmos. 93(D12):15879–88. doi:10.1029/JD093iD12p15879
- Main, H., S. Hurwitt, and P. Roberts. 1999. Spatial and temporal characteristics of California PAMS and long-term trend site VOC data (1990–1997). Report STI-9982-1883-FR for the Environmental Protection Agency. Petaluma, CA: Sonoma Technology, Inc.
- McDonald, B.C., T.R. Dallman, E.W. Martin, and R. Harley. 2012. Long-term trends in nitrogen-oxide emissions from motor vehicles at national, state, and air basin scales. J. Geophys. Res. Atmos. 117: D00V18. doi:10.1029/2012JD018304
- McDonald, B., D. Gentner, A. Goldstein, and R. Harley. 2013. Long term trends in motor vehicle emissions in US urban areas. Environ. Sci. Technol. 47:10022–31. doi:10.1021/es401034z
- Midwest Ozone Group. 2013. Air trends project. http://midwestozonegroup.com/AirTrendsJuly2013Public.html (accessed December 8, 2014).
- NARSTO. 2000. An assessment of tropospheric ozone pollution: A North American perspective. NARSTO Report 1000040. Palo Alto, CA: Electric Power Research Institute.
- National Research Council. 1991. Rethinking the ozone problem in urban and regional pollution. Washington, DC: National Academies Press.
- Oltmans, S., A. Lefohn, J. Harris, I. Galbally, H. Scheel, G. Bodeker, E. Brunke, H. Claude, D. Tarasick, B. Johnson, P. Simmonds, D. Shadwick, K. Anlauf, K. Hayden, F. Schmidlin, T. Fujimoto, K. Akagi, S. Meyer, S. Nichol, J. Davies, A. Redondas, and E. Cuevas. 2006. Long-term changes in tropospheric ozone. Atmos. Environ. 40:3156–73. doi:10.1016/j.atmosenv.2006.01.029
- Oltmans, S., A. Lefohn, J. Harris, and D. Shadwick. 2008. Background ozone levels of air entering the west coast of the US and assessment of longer-term changes. Atmos. Environ. 42:6020–38. doi:10.1016/j.atmosenv.2008.03.034
- Oltmans, S., A. Lefohn, J. Harris, D. Tarasick, A. Thompson, H. Wernli, B. Johnson, J. Daview, P. Novelli, P. Montzka, C. Sweeney, I. Patrick, A. Jefferson, A. Dann, T. Ray, M. Shapiro, and B. Holben. 2010. Enhanced ozone over western North America from biomass burning in Eurasia during April 2008 as seen in surface and profile observations. Atmos. Environ. 44:4497–509. doi:10.1016/j.atmosenv.2010.07.004
- Oltmans, S., A. Lefohn, D. Shadwick, J. Harris, H. Scheel, I. Galbally, D. Tarasick, B. Johnson, E. Brunke, H. Claude, G. Zeng, S. Nichol, F. Schmidlin, J. Davies, E. Cuevas, A. Redondas, H. Naoe, T. Kakano, and T. Kawasato. 2013. Recent tropospheric ozone changes—A pattern dominated by slow or no growth. Atmos. Environ. 67:331–51. doi:10.1016/j.atmosenv.2012.10.057
- Parrish, D., and F. Fehsenfeld. 2000. Methods of gas phase measurements of ozone, ozone precursors and aerosol precursors. Atmos. Environ. 34:1921–58. doi:10.1016/S1352-2310(99)00454-9
- Parrish, D., D. Miller, and A. Goldstein. 2009. Increasing ozone in marine boundary layer inflow at the west coast of North America and Europe. Atmos. Chem. Phys. 9:1303–23. doi:10.5194/acp-9-1303-2009
- Parrish, D., and W. Stockwell. 2015. Urbanization and air pollution. Then and now. Earth Space News 96:10–15. doi:10.1029/2015EO021803
- Pollack, I., T. Ryerson, M. Trainer, J. Neuman, J. Roberts, and D. Parrish. 2013. Trends in ozone its precursors and related secondary oxidation products in Los Angeles, California: A synthesis of measurements from 1960–2010. J. Geophys. Res. Atmos. 118:5891–911. doi:10.1002/jgrd.50472
- Reynolds, S., C. Blanchard, and S. Ziman. 2003. Understanding the effectiveness of precursor reductions in lowering 8-hour ozone concentrations. J. Air Waste Manage. Assoc. 53:195–205. doi:10.1080/10473289.2003.10466143
- Reynolds, S., C. Blanchard, and S. Ziman. 2004. Understanding the effectiveness of precursor reductions in lowering the 8-hour O3 concentration. Part II—Eastern United States. J. Air Waste Manage. Assoc. 54:1452–70. doi:10.1080/10473289.2004.10471003
- Ridley, B. 1991. Recent measurements of oxidized nitrogen compounds in the troposphere. Atmos. Environ. 25A:1905–26. doi:10.1016/0960-1686(91)90273-A
- Sather, M., and K. Cavendar. 2007. Trends analysis of ambient 8-hour ozone and precursor monitoring data in the south central United States. J. Environ. Monit. 9:143–50. doi:10.1039/b617603h
- Sather, M., and K. Cavendar. 2012. Update of long-term trends analysis of ambient 8-hour ozone and precursor monitoring data in the South Central U.S.; Encouraging news. J. Environ. Monit. 14:666–76. doi:10.1039/c2em10862c
- Seinfeld, J., and S. Pandis. 1998. Atmospheric chemistry and physics: From air pollution to climate change. New York, NY: Wiley-Interscience. 1998.
- Sillman, S. 1999. The relation between ozone, NOx and hydrocarbons in urban and polluted rural environments. Atmos. Environ. 33:1821–45. doi:10.1016/S1352-2310(98)00345-8
- Sillman, S., J. Logan, and S. Wofsy. 1990. The sensitivity of ozone to nitrogen oxides and hydrocarbons in regional ozone episodes. J. Geophys. Res. Atmos. 95:1837–51. doi;10.1029/JD095iD02p01837
- Simon, H., A. Reff, B. Wells, and N. Frank. 2015. Ozone trends across the United States over a period of decreasing NOx and VOC emissions. Environ. Sci. Technol. 49:186–95. doi:10.1021/es504514z
- Southeastern Michigan Council of Governments (SEMCOG). 2010. Ozone air quality trends. www.semcog.org/workArea/DownloadAssetaspx?id=424970582 (accessed January 2015).
- Texas Commission on Environmental Quality (TCEQ). 2013. Air quality successes—Emission inventory and Air quality successes—Criteria pollutants. http://www.tceq.texas/air quality/air success/air-success… ( accessed October 2103.)
- Thornton, J., P. Wooldridge, R. Cohen, M. Martinez, H. Harder, W. Brune, E. Williams, J. Roberts, F. Fehsenfeld, F. Hall, S. Shetter, B. Wert, and A. Fried. 2002. Ozone production rates as a function of NOx abundances and HOx production rates in the Nashville urban plume. J. Geophys. Res. 107:4146, doi:10.1029/2001JD000932
- Trainer, M., D.D. Parrish, M.P. Buhr, R. Norton, F. Fehsenfeld, K. Anlauf, J. Bottenheim, Y. Tang, H. Weibe, J. Roberts, R. Tanner, L. Newman, V. Bowersox, J. Meagher, K. Olszyna, M. Rodgers, T. Wang, H. Berresheim, K. Demerjian, and U. Roychowdhury. 1993. Correlation of ozone with NOy in photochemically aged air. J. Geophys. Res. Atmos. 98(D2):2917–25. doi:10.1029/92JD01910
- Trainer, M., D. Parrish, P. Coldan, J. Roberts, and F. Fehsenfeld. 2000. Review of observation-based analysis of regional factors influencing ozone concentration. Atmos. Environ. 34:2045–62. doi:10.1016/S1352-2310(99)00459-8
- U.S. Bureau of the Census. 2015. Metropolitan and micropolitan statistical areas of the United States and Puerto Rico. http://www2.census.gov/geo/maps/metroarea/us_wall/Dec_2009/cbsa_us_1209_large.gif (accessed January 2015).
- Vingarzan, R. 2004. A review of ozone background levels and trends. Atmos. Environ. 38:3431–42. doi:10.1016/j.atmosenv.2004.03.030
- Walsh, K., M. Milligan, M. Woodman, and J. Sherwell. 2008. Data mining to characterize ozone behavior in Baltimore and Washington, DC. Atmos. Environ. 42:4280–92. doi:10.1016/j.atmosenv.2008.01.012
- Wolff, G., A. Dunker, P. Porter, and I. Zurbenko 2001. Ozone air quality over North America: Part 1—A review of reported trends. J. Air Waste Manage. Assoc. 51:273–82. doi:10.1080/10473289.2001.10464270
- Xing, J., J. Pleim, R. Mathur, G. Pouliot, C. Hogrefe, C.-M. Gan, and C. Wei. 2013. Historical gaseous and primary aerosol emissions in the United States from 1990 to 2010. Atmos. Chem. Phys. 13:7531–49. doi:10.5194/acp-13-7531-2013
- Xing, J., R. Mathur, J. Pleim, C. Hogrefe, C.-M. Gan, D. Wong, C. Wei, R. Gilliam, and G. Pouliot. 2015. Observations and modeling of air quality trends over 1990–2010 across the Northern Hemisphere: China, the United States and Europe. Atmos. Chem. Phys. 15:2723–47. doi:10.5194/acp-15-2723-2015
- Zhou, W., D. Cohan, and S. Napelenok. 2013. Reconciling NOx emissions reductions and ozone trends in the U.S., 2002–2006. Atmos. Environ. 70:236–44. doi:10.1016/j.atmosenv.2012.12.038