
Abstract
The carbohydrate-active enzyme (CAZyme) genes of Trametes contribute to polysaccharide degradation. However, the comprehensive analysis of the composition of CAZymes and the biosynthetic gene clusters (BGCs) of Trametes remain unclear. Here, we conducted comparative analysis, detected the CAZyme genes, and predicted the BGCs for nine Trametes strains. Among the 82,053 homologous clusters obtained for Trametes, we identified 8518 core genes, 60,441 accessory genes, and 13,094 specific genes. A large proportion of CAZyme genes were cataloged into glycoside hydrolases, glycosyltransferases, and carbohydrate esterases. The predicted BGCs of Trametes were divided into six strategies, and the nine Trametes strains harbored 47.78 BGCs on average. Our study revealed that Trametes exhibits an open pan-genome structure. These findings provide insights into the genetic diversity and explored the synthetic biology of secondary metabolite production for Trametes.
1. Introduction
Trametes, one of white-rot basidiomycetes, belongs to the family Polyporaceae and the class Agaricomycetes of fungi, commonly grows in the tiled layer of decaying wood [Citation1], and uses deciduous tree [Citation2]. This genome currently comprises at least 20 species until April 2018 [Citation3,Citation4], such as Trametes sp., Trametes trogii, and Trametes villosa CCMB561. Trametes is known to decolorize dyes [Citation1], and several of its species have been used for centuries in traditional medicine in East Asia countries [Citation5]. Trametes sp. SQ01 can decolorize azo, anthraquinone, and triphenylmethane dyes [Citation6]. T. trogii, a worldwide-distributed white-rot fungi and an outstanding laccase producer, degrades all lignocellulosic materials [Citation7]. T. villosa CCMB561, a common species in the Brazilian semiarid region [Citation8], has been isolated from decaying wood and is considered a high-potential fungus for biotechnological applications because it is a good producer of the three important ligninolytic enzymes (laccase, manganese peroxidase, and lignin peroxidase) [Citation4]. The laccases from Trametes can be used as a biocatalyst in enzymatic biofuel cells [Citation9,Citation10]. Hence, understanding the genetic diversity of Trametes is beneficial for its biotechnological applications.
Carbohydrate-active enzymes (CAZymes) are involved in the polysaccharide degradation of plant cell wall [Citation11], lignin, cellulose, hemicellulose, and pectin [Citation12]. In particular, CAZyme classes (glycoside hydrolases [GH], carbohydrate esterases [CEs], and polysaccharide lyases) play central roles in plant biomass decomposition by bacteria and fungi [Citation13]. The total expression levels of CAZyme genes of T. versicolor F21a are almost twice as that of the control group during algaecide, and the majority of expressed CAZyme genes belong to GH family and auxiliary activities [Citation14]. Similarly, 590 genes of T. villosa CCMB561 are identified as CAZyme genes [Citation4]. Although the compositions of CAZyme genes of fewer species of Trametes have been reported, the composition of CAZyme genes of Trametes has not been comprehensively analyzed.
Secondary metabolites (referred to as natural products or specialized metabolites) are the foundation of many drugs [Citation15] and important chemicals used in agriculture and nutrition [Citation16]. In general, fungi are rich sources of thousands of second metabolites, and fungal second metabolites can be grouped into four primary chemical types: polyketides (PKS), terpenoids, shikimin acid-derived compounds, and non-ribosomal peptides (NRPS) [Citation17]. The secondary metabolites of fungi are crucial in its development and actively shape interactions with other microbes [Citation18]. The number of genes coded for the secondary metabolism of ascomycetes is higher than that of basidiomycetes, archeo-ascomycetes, and chytridiomycetes [Citation19]. Several fungal genomes have been sequenced, and the development of various genome mining software tools, such as antiSMASH [Citation16] and ClusterFinder [Citation20], enables researchers to analyze the biosynthetic gene clusters (BGCs) of secondary metabolites. Genomes of filamentous fungi contain up to 90 potential BGCs encoding their diverse secondary metabolites [Citation21], and 24 genomes of Penicillium are mined for BGCs and associated PKS and NRPS BGCs to known pathways [Citation22]. However, no systemically comparative analysis has been conducted to identify the BGCs of secondary metabolite in Trametes.
In this study, we collected nine available genomic data from eight species of Trametes and conducted a comparative genomic analysis to investigate the genetic diversity and the synthetic biology of secondary metabolites. We obtained the Trametes pan-genome, annotated the pan-genome against clusters of orthologous group (COG) database and eggNOG database, and detected CAZyme genes. The BGCs for nine Trametes strains were also predicted. Our results showed that Trametes exhibits an open pan-genome structure and diverse genetic diversity for Trametes strains. The different distributions of CAZymes of Trametes reveal variations in carbohydrate utilization for Trametes strains. The predicted BGCs with unknown functions in Trametes suggested that Trametes have a great potential value for secondary metabolite production.
2. Materials and methods
2.1. Genomic data collection of Trametes
We used “Trametes” as the keyword and searched in genome database of National Center for Biotechnology Information (NCBI) on September 14, 2018. We found that Trametes comprising 8 species and containing nine full genomes, such as T. cinnabarina BRFM137, T. cinnabarina FP104138-Sp, T. coccinea BRFM310, T. hirsuta, T. polyzona, T. pubescens, T. sp. AH28-2, T. versicolor FP-101664SS1, and T. villosa. Although there are 20 species of Trametes were discovered, there are only nine full genomes affiliated with 8 species can be obtained in public database. As such, we downloaded the genomic data (draft) of these strains for comparative genomics analysis.
2.2. Gene prediction and ortholog identification
We conducted the genome annotation for nine strains using Prodigal version 2.6 [Citation23]. In particular, to predict the genes and proteins for nine genomic data of Trametes, we applied the Prodigal with default setting to recognize open reading frames (ORFs) and protein sequences [Citation24]. We identified the protein orthologs of Trametes by using OrthMCL version 2.0.9 [Citation25] with e-value <1e-5 and inflation parameter of 1.5. We divided the homologous clusters into three groups: core, accessory and specific groups. The core genes or proteins represent the genes or proteins shared in all nine genomes of Trametes used in our study, while the accessory genes or proteins comprised genes or proteins shared by at least two strains but not all nine Trametes strains. The rest of genes or proteins only occurred in one strain were clustered into specific groups (strain-specific genes or proteins). We obtained the protein sequences of homologous clusters for Trametes.
2.3. Phylogeny analysis
The nuclear ribosomal ITS region has been reported as the primary fungal barcode marker [Citation26] and ITS1 or ITS2 was widely used to identify the broadest range of fungi [Citation27]. Hence, we applied ITSx [Citation28], which is an open source software utility to extract the highly variable ITS1 and ITS2 subregions from ITS sequences, to extract the ITS1 and ITS2 sequences from the genomic data of nine Trametes strains. Meanwhile, we chose the ITS1 and ITS2 sequences of Coriolopsis caperata (accession number: AB158316) as outgroup. In our work, due to the fact that the ITS1 and ITS2 sequences of Trametes coccinea were not extracted, we selected the ITS1 and ITS2 sequences of Pycnoporus coccineus strain MUCL 38523 [Citation29] (one strain of T. coccinea, accession number: FJ873395) to represent T. coccinea and construct the phylogenetic tree. The ITS1 and ITS2 sequences were aligned using the MUSCLE version 3.8.31 [Citation30] with default setting, respectively. The multiple sequences alignment result was eliminated by using Gblocks version 0.91b [Citation31] to remove the regions that were divergent, misaligned, or with a larger number of gaps. Then we used PHYLIP version 3.696 [Citation32] with 100 bootstrap iterations to construct a maximum-likelihood tree based on the concatenated multiple sequences alignment result. Moreover, we built a pan-genome tree by MEGA software version 5 [Citation33] based on the composition of orthologous. In particular, we calculated the pairwise Manhattan distance between each strain based on the presence and absence of orthologs proteins.
2.4. Functional annotation
To obtain the COG functions, the protein sequences of homologous clusters for Trametes were aligned to individual COG proteins [Citation34], which downloaded from NCBI (https://www.ncbi.nlm.nih.gov/COG/) using BLSATP with e-value <1e-4 and the top one of the results was chosen as the best annotation of each protein. The annotations of the protein sequences of homologous clusters were assigned to 25 functional categories. We also annotated the protein sequences of homologous clusters against eggNOG annotation database by using the eggnog-mapper [Citation35] and we divided the GO annotations of protein sequences of homologous clusters into three groups, including biological process, molecular function, and cellular component. We annotated these protein sequences against CAZyme database to investigate the functional composition related to carbohydrate metabolism. In particular, we downloaded the CAZy database from dbCAN [Citation36] (http://csbl.bmb.uga.edu/dbCAN/). According to the manual of dbCAN CAZyme annotation, protein sequences were annotated by running hmmscan from the HMMER version 3.1b1 [Citation37,Citation38] with default setting and the annotated results were summarized into GT, GH, carbohydrate-binding module (CBM), AA, PL, and CE.
2.5. BGC detection
To detect the biosynthetic pathways of secondary metabolites for Trametes, antiSMASH version 4.0 [Citation16] was used to identify the BGCs. A set of nine genome sequences of Trametes was used as the input data for predicting the BGCs by using antiSMASH with the mode of fungi and other default setting. The BGCs were determined to their subtypes (e.g., type I polyketide, terpenoid).
3. Results and discussion
3.1. Pan-genome construction and analysis
Nine available genomic data from eight species of Trametes were collected for the pan-genome analysis. The assembled genome of Trametes ranged from 31.62 Mb to 57.98 Mb, and the number of contigs/scaffolds ranged from 13 to 10,327. In addition, the number of CDSs/ORFs predicted by Prodigal ranged from 56,735 to 101,817 (). The number of genes predicted in our study for Trametes was greater than that in a previous report. The predicted genes of T. villosa CCMB561 in the present study were 101,817, whereas those in the previous study were 16,711 [Citation4]. This difference might be due owing to the different tools and parameters. Orthologs from the 610,421 high-quality proteins of Trametes were also detected to characterize the differences of genomic composition among the nine strains. A total of 82,053 homologous clusters were obtained, and the accumulation curve of the homologous cluster showed that Trametes exhibits an open pan-genome structure (). The size of Trametes pan-genome tended to increase gradually with new strains and comprised 82,053 non-redundant genes within nine strains (). On the contrary, the size of core-genome tended to decrease progressively with new strains and comprised 8518 non-redundant genes within the nine Trametes strains ().
Figure 1. Genetic diversity of strains in Trametes. (a) Size of pan-genome (red) and core genome (blue) shared by different genomes, respectively; (b) Core and specific genes families of nine Trametes strains. Number of core genome shared by all strains is in the center (8518). Number of non-overlapping portions of each oval represents the size of specific families; (c) Phylogenetic tree based on the ITS1 sequences of nine Trametes strains and Coriolopsis caperata; (d) Phylogenetic tree based on the ITS2 sequences of nine Trametes strains and C. caperata. Coriolopsis caperata was selected as an outgroup to root the topology of phylogenetic tree; (e) UPGMA tree of Manhattan distance based on the pan-genome composition of nine Trametes strains.
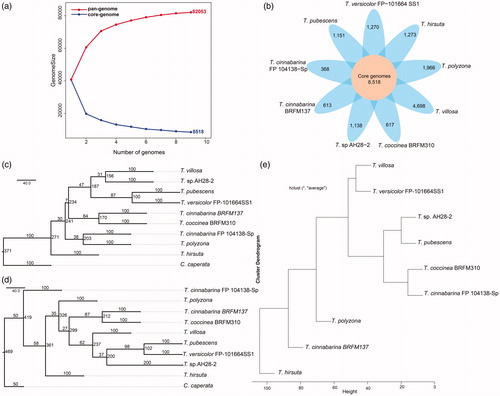
Table 1. Comparison of the genomic features of nine strains in Trametes genus.
A total of 60,441 accessory genes and 13,094 specific genes of Trametes were obtained. The distribution of accessory genes varied from 17,587 to 31,856. Although T. cinnabarina FP104138-Sp was the smallest () among the nine Trametes strains, it had the maximum number (31,856) of accessory genes. Meanwhile, T. hirsuta had the minimum number (17,587) of accessory genes. The number of specific orthologs of the nine Trametes strains ranged from 368 to 4698. Trametes villosa CCMB561 possessed the highest number (4,698) of specific genes, whereas T. cinnabarina FP 104138-Sp had the minimum number (368) of specific genes (). The distribution of accessory and specific genes revealed that Trametes genomes are considerably diverse.
3.2. Phylogenetic analysis of Trametes pan-genome
The nuclear ribosomal internal transcribed spacer (ITS) region was the primary fungal barcode marker [Citation26]. In addition, ITS was useful in identifying the broadest range of fungi [Citation27]. A phylogenetic tree for Trametes and C. caperata, which was selected as an outgroup to root the topology of the tree, was built based on their ITS1 and ITS2 sequences, respectively, to analyze the phylogenetic relationships for the nine Trametes strains. The ITS1 and ITS2 sequences had a strong power to differentiate the nine Trametes strains (). Among the phylogenetic trees, T. pubescens, T. versicolor FP-101664 SS1, and T. sp. AH28-2 had a close evolutionary relationship, and T. cinnabarina BRFM137 had a close evolutionary relationship with T. coccinea BRFM310. However, the evolutionary relationship of T. hirsuta was distant from that of the remaining strains of Trametes ().
Another phylogenetic tree was built based on the presence and absence of genes in pan-genome to gain insights into the evolutionary relationships among the strains of Trametes. The results showed that T. villosa had a close evolutionary relationship with T. versicolor FP-101664 SS1. Meanwhile, T. sp.AH28-2, T. pubescens, T. coccinea BRFM310, and T. cinnabarina FP 104138-Sp had a close evolutionary relationship with one another (), revealing that they had similar genetic composition and functional metabolism. In addition, the evolutionary relationship of T. hirsuta was distant from the rest of Trametes strains (), suggesting that the functional composition of T. hirsuta was different from that of the remaining Trametes strains, and the functional metabolism of Trametes was highly diverse.
3.3. Functional characterization of the Trametes pan-genome
The Trametes pan-genome against the COG database was annotated to characterize the functions of the accessory clusters, core clusters, and specific genes and to investigate the functional diversity of the nine Trametes strains. The orthologous genes of the pan-genome were primarily dominant in “general function prediction only” (R, 1,758, 15.47%); “replication, recombination, and repair” (L, 998, 8.78%); “amino acid transport and metabolism” (E, 996, 8.77%); “translation, ribosomal structure, and biogenesis” (J, 755, 6.64%); and “carbohydrate transport and metabolism” (G, 753, 6.63%, ). Moreover, Trametes had 442 (3.89%) orthologous genes assigned to “secondary metabolite biosynthesis, transport, and catabolism” (Q, ). Similarly, the accessory genome was enriched in “general function prediction only” (R, 1,411, 15.29%); “replication, recombination, and repair” (L, 810, 8.78%); and “amino acid transport and metabolism” (E, 799, 8.66%, ). In addition, the core gene clusters were predominantly composed of genes involved in “general function prediction only” (R, 88, 20.09%), “energy production and conversion” (C, 51, 11.64%), “amino acid transport and metabolism” (E, 38, 8.68%), and “carbohydrate transport and metabolism” (G, 36, 8.21%, ). Finally, specific genes were primarily contributed to the functions involved in “replication, recombination, and repair” (L, 610, 28.44%) and “general function prediction only” (R, 259, 12.07%, ). The high proportions of accessory genes (9230, 81.23%) and specific genes (2145, 18.88%) in each strain of Trametes might lead to diverse functions.
Figure 2. COG and GO annotation of gene families. (a) Distribution of COG annotation of pan-genome that assigned in 25 functional categories; (b) Distribution of GO annotation of pan-genome in biological process, molecular function and cellular component. Accessory clusters (pink), core clusters (green), and specific genes (wathet).
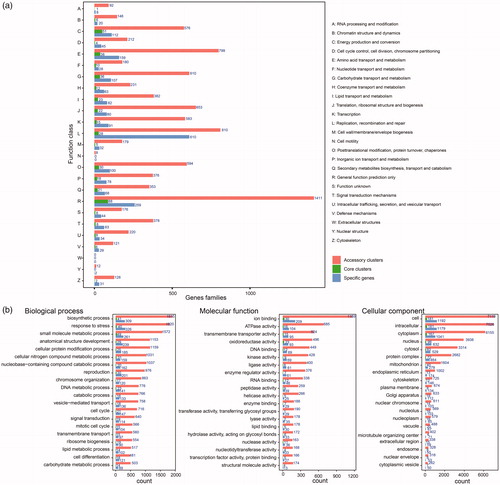
Gene Ontology (GO) analysis was conducted to characterize the genetic functions of the pan-genome and explore the evolution of function catalogs. These genes were categorized on the basis of the biological process, cellular component, and molecular function. The results showed that a great number of genes of pan-genome were involved in various enzymes, metabolic pathways, and biological processes. In particular, enrichment analysis of biological processes showed that many genes were classified into the following three categories: biosynthetic process (2265), response to stress (2186), and small molecule metabolic process (1882, ). In addition, 606 orthologous genes contributed to the carbohydrate metabolism (). The classification based on the molecular function showed that the majority of genes were grouped into binding (such as ion binding, DNA binding, and RNA binding) and enzymes (such as ATPase activity, oxidoreductase activity, and kinase activity). However, various genes were also enriched in other categories, such as transporter activity and enzyme regulator activity (). Enrichment analysis of cellular component showed that high proportions of genes were grouped on the basis of their functions, such as cell (8492), intracellular (8386), and cytoplasm (7363, ). Among these categories, accessory clusters were highly represented in these functions than core and specific genes (). Therefore, the diversity of accessory clusters might influence their functional diversity, allowing these strains to utilize the resources of the surrounding environment and adapt to the environment.
3.4. Identification of CAZymes for Trametes
White-rot basidiomycetes had the ability to decompose lignin most efficiently [Citation48], and fungi were CAZymes and lignin-degrading enzymes for the degradation of lignocellulose [Citation49]. Trametes, as one of the branches of white-rot basidiomycetes, and its members, such as T. villosa [Citation48] and T. gibbosa BRFM 952 [Citation50], had been reported that these strains had an unexpected high activity on lignin or crystalline cellulose. A previous study had shown that CAZymes played a central role in the degradation of glycoconjugate, oligosaccharide, and polysaccharide [Citation37]. Hence, we systemically identified the Trametes pan-genome against the carbohydrate-active enzymes database (CAZy database) and grouped CAZyme genes into different CAZyme families and carbohydrate-binding modules (CBMs) on the basis of family-specific Hidden Markov Models (HMMs) to obtain systematic understanding of CAZymes of Trametes [Citation51]. A total of 280 orthologous genes () were identified and grouped into 87 CAZyme families. A large proportion of orthologous genes were cataloged into GHs (35.36%), glycosyltransferases (21.07%, GTs), and CEs (13.57%, ). GH hydrolyzed the glycosidic bond between two or more carbohydrates [Citation37], contributed most of the catalytic enzymes, and was involved in lignocellulosic degradation [Citation52]. A previous study identified 237 GHs, 78 GTs, 12 polysaccharide lyase (PLs), 69 CEs, and 112 auxiliary activity (AAs) in T. villosa CCMB561 [Citation4]. Moreover, the number of CAZymes of accessory clusters was higher than that of core clusters and specific genes (), and the number of genes belonging to different CAZymes in each Trametes strain () was different. This finding suggested a remarkable difference in the ability of strains of Trametes for carbohydrate degradation.
Figure 3. Distributions of CAZymes in nine Trametes strains of Trametes. (a) Distribution of CAZymes in Trametes pan-genome; (b) Distribution of CAZymes of pan-genome in each Trametes strain grouped by accessory clusters, core clusters and specific genes. The numbers shown in subgraph represents the orthologous genes assigned by CAZymes and grouped by accessory clusters (pink), core clusters (green), and specific genes (wathet); (c) Distribution of CAZymes of accessory clusters in each Trametes strain; (d) Distribution of CAZymes of core clusters in each Trametes strain; (e) Distribution of CAZymes of specific genes in each Trametes strain. The numbers shown in subgraphs represent the orthologous genes assigned by CAZymes. GT: glycosyltransferase; GH: glycoside hydrolase; CE: carbohydrate esterase; CBM: carbohydrate-binding molecule; AA: auxiliary activities; PL: polysaccharide lyases.
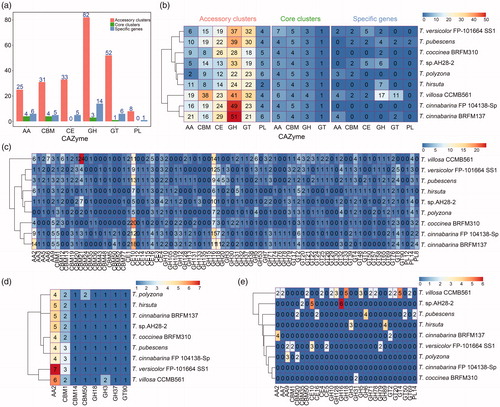
The 87 CAZyme families were subdivided on the basis of accessory clusters, core clusters, and specific genes to gain insights into the CAZyme composition of the nine Trametes strains (). For the orthologous genes of accessory clusters, a slice of CAZyme families, such as AA9, CBM1, CE10, CE16, CE4, GH10, GH16, GH18, GH5, GH79, GT2, GT20, GT76, and PL14, were detected in all nine Trametes strains (). Among these CAZyme families, the number of CE10 (ranging from 5 to 20) and GH16 (ranging from 6 to 15) were higher than other CAZyme families (). A previous study reported that CE10 family includes various esterases [Citation36]. GHs could hydrolyze glycosidic bonds among carbohydrates or between a carbohydrate and a non-carbohydrate moiety [Citation36]. In addition, GH16 comprises a large of glycosidases and transglycosidases, which catalyzed a range of terrestrial and marine polysaccharides [Citation53]. The counts of AA2 of T. cinnabarina BRFM137 (14) and CBM21 of T. villosa CCMB561 (24) was higher than that of other strains (). By contrast, the distributions of CAZyme families of core clusters, such as AA2, CBM1, CBM14, CBM50, GH18, GH3, GH37, and GT90, in Trametes strains were similar (). Moreover, the orthologous genes of specific genes were assigned to different CAZyme families (). For example, 14 CAZyme families, including GH18 and GT4, were dominant in T. villosa CCMB561, and CE10 and GH16 were enriched in T. sp. AH28-2. Notably, AA7 was only detected in T. villosa CCMB561; CE5 was only identified in T. versicolor FP-101664 SS1; CH6 only appeared in T. pubescens; and CH89 was only detected in T. hirsuta. The unique CAzyme family and the differential composition of CAZymes in Trametes revealed their distinct ability to metabolize carbohydrates. In summary, the composition of CAZyme families of the nine Trametes strains showed that these strains had the ability to metabolize the same carbohydrates and to degrade different carbohydrates.
3.5. Identification of Trametes BGCs
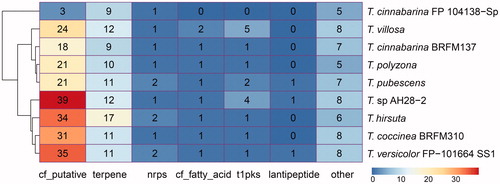
AntiSMASH was applied in predicting BGCs to further understand the secondary metabolite of Trametes. Our results showed that the BGCs of Trametes were primarily divided into six strategies, such as putative cluster of unknown type identified with the ClusterFinder algorithm (cf_putative), terpene cluster (terpene), nonribosomal peptide synthetase cluster (nrps), putative fatty acid cluster identified with the ClusterFinder algorithm (cf_fatty_acid), type I PKS cluster (t1pks), and lanthipeptide cluster (lantipeptide, ). In particular, the nine Trametes strains harbored 47.78 BGCs on average, and the number of BGCs of Trametes ranged from 18 (T. cinnabarina FP 104138-Sp) to 66 (T. sp. AH28-2, ). Clusters of cf_putative and terpenes represented the majority of predicted specialized metabolite BGCs and contributed to the highest share among the nine Trametes strains (). The results showed that the clusters of cf_putative were enriched in the nine Trametes strains (ranging from 3 to 39 with an average of 25 BGCs, ), whereas T. cinnabarina FP 104138-Sp harbored only three clusters of cf_putative. When comparing the BGCs of terpene, T. hirsuta harbored 17 BGCs, and T. cinnabarina BRFM137 and T. cinnabarina FP 104138-Sp all harbored nine BGCs. Moreover, the majority of these identified terpene BGCs could not be linked to known terpene BGCs. In addition, many predicted BGCs were grouped into “other” of these nine strains.
Figure 4. Distribution of biosynthetic gene clusters in nine Trametes strains of Trametes. The distribution of biosynthetic gene clusters in each Trametes strain. The numbers shown in graph represent the counts of different types of biosynthetic gene clusters predicted by antiSMASH for each Trametes strain. Note: cf_putative, putative cluster of unknown type identified with the ClusterFinder algorithm; terpene, terpene cluster; nrps, nonribosomal peptide synthetase cluster; cf_fatty_acid, putative fatty acid cluster identified with the ClusterFinder algorithm; t1pks, type I PKS cluster; lantipeptide, lanthipeptide cluster; other, cluster containing a secondary metabolite-related protein that does not fit into any other category.
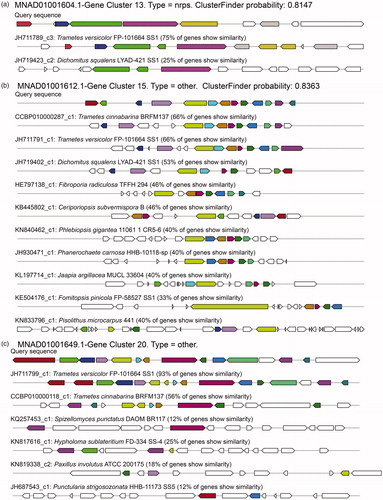
Although the secondary metabolites of predicted BGCs were unknown, we identified a few enzymes as the core biosynthetic enzymes, such as cembrene C synthase, pristinol synthase, (+)-epi-cubebol synthase, and epi-isozizaene synthase, in several BGCs in most strains of Trametes. To gain insights into the BGCs of Trametes, three BGCs of T. pubescens were chosen as examples (). In particular, the BGCs of No. 13 were detected by the ClusterFinder algorithm built-in antiSMASH with 81.47% probability, and the homologous gene clusters were identified from T. versicolor FP-101664 SS1 and Dichomitus squalens LYAD-421 SS1 with 75% and 25%, respectively, of genes showing similarity (). In addition, the monomer of the predicted core structure of the substrate was ala-ala (C6H12O2N2). The BGCs of No. 15 was detected as other unknown cluster with 83.63% probability, and the homologous gene clusters were identified from T. cinnabarina BRFM137 and T. versicolor FP-101664 SS1 that both had 66% of genes showing similarity (), whereas the BGCs of No. 20 was also detected as other unknown cluster, and the homologous gene clusters were identified from T. versicolor FP-101664 SS1 and T. cinnabarina BRFM 137 with 93% and 56%, respectively, of genes showing similarity (). In summary, these results revealed that many BGCs of Trametes were undiscovered, and Trametes had a potential value for producing secondary metabolites.
Figure 5. Probability and homologous gene clusters for three putative BGCs of T. pubescens. The probability and homologous gene clusters for the putative BGCs of (a) no. 13; (b) no. 15; (c) no. 20. In the subgraph, the same colors of the boxes with arrows are similar genes.
4. Conclusion
In conclusion, we systemically comparative analyzed the Trametes pan-genome, one of important branches of white-rot basidiomycetes, based on the available genome of nine Trametes strains and we explored the functional composition of nine Trametes strains, including COG annotation, GO annotation, and CAZyme families. Furthermore, we predicted the BGCs for each strain of Trametes. Our comparative analysis revealed that Trametes exhibited an open pan-genome structure and tremendous diversity in both the number and the variety of genes. The results of comparative analysis of Trametes pan-genome also showed the differences in the composition of functional metabolisms and the evolutionary relationships among the nine Trametes strains. In addition, we observed that the high proportion of accessory genes and specific genes annotated with different COG annotations and GO annotations showed that the diversity of the functions among the Trametes strains of Trametes. Importantly, our results showed that the CAZyme families, such as GH, GT, and CE, enriched in the Trametes strains, while the number of CAZymes of accessory clusters is higher than that of core clusters and specific genes and the number of genes belonged to different CAZymes in each Trametes strain suggested that the potential ability to degrade different carbohydrates for different strains of Trametes. Finally, we predicted many BGCs with unknown functions in Trametes, which suggested that Trametes has a great potential value for producing secondary metabolite. Together, we uncover the genetic diversity and synthetic biology of secondary metabolite production of Trametes in this work. Our results provide a broader understanding of Trametes and importantly shed the lights on the composition of CAZyme families and the BGCs of Trametes.
Authors’ contributions
This study was designed by YZ and ZDX. YZ collected and downloaded the data. YZ and YJC analyzed the data. YZ, JJW, and ZDX wrote the initial draft of the manuscript. All authors revised the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Cho KS, Ryu HW. Biodecolorization and biodegradation of dye by fungi: a review. KSBB J. 2015;30:203–222.
- Ryu H, Ryu HW, Cho KS. Characterization of dye decolorization in cell-free culture broth of Trametes versicolor CBR43. J Microbiol Biotechnol. 2017;27:155–160.
- Justo A, Miettinen O, Floudas D, et al. A revised family-level classification of the Polyporales (Basidiomycota). Fungal Biol. 2017;121:798–824.
- Ferreira DSS, Kato RB, Miranda FM, et al. Draft genome sequence of Trametes villosa (Sw.) Kreisel CCMB561, a tropical white-rot Basidiomycota from the semiarid region of Brazil. Data Brief. 2018;18:1581–1587.
- Knežević A, Stajić M, Sofrenić I, et al. Antioxidative, antifungal, cytotoxic and antineurodegenerative activity of selected Trametes species from Serbia. PLoS One. 2018;13:e0203064.
- Yang XQ, Zhao XX, Liu CY, et al. Decolorization of azo, triphenylmethane and anthraquinone dyes by a newly isolated Trametes sp. SQ01 and its laccase. Process Biochem. 2009;44:1185–1189.
- Levin L, Herrmann C, Papinutti VL. Optimization of lignocellulolytic enzyme production by the white-rot fungus Trametes trogii in solid-state fermentation using response surface methodology. Biochem Eng J. 2008;39:207–214.
- Neves M, Baseia I, Drechsler-Santos E, et al. Guide to the common fungi of the semiarid region of Brazil. Florianópolis: TECC Editora; 2013.
- Milton RD, Giroud F, Thumser AE, et al. Hydrogen peroxide produced by glucose oxidase affects the performance of laccase cathodes in glucose/oxygen fuel cells: FAD-dependent glucose dehydrogenase as a replacement. Phys Chem Chem Phys. 2013;15:19371–19379.
- Salaj-Kosla U, Pöller S, Schuhmann W, et al. Direct electron transfer of Trametes hirsuta laccase adsorbed at unmodified nanoporous gold electrodes. Bioelectrochemistry. 2013;91:15–20.
- Davies GJ, Williams SJ. Carbohydrate-active enzymes: sequences, shapes, contortions and cells. Biochem Soc Trans. 2016;44:79–87.
- Sista Kameshwar AK, Qin W. Comparative study of genome-wide plant biomass-degrading CAZymes in white rot, brown rot and soft rot fungi. Mycology. 2018;9:93–105.
- Zhao Z, Liu H, Wang C, et al. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics. 2013;14:274.
- Dai W, Chen X, Wang X, et al. The Algicidal Fungus Trametes versicolor F21a eliminating blue algae via genes encoding degradation enzymes and metabolic pathways revealed by transcriptomic analysis. Front Microbiol. 2018;9:826.
- Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–335.
- Blin K, Wolf T, Chevrette MG, et al. antiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017;45:W36–W41.
- Pusztahelyi T, Holb IJ, Pócsi I. Secondary metabolites in fungus–plant interactions. Front Plant Sci. 2015;6:573.
- Keller NP. Fungal secondary metabolism: regulation, function and drug discovery. Nat Rev Microbiol. 2018; 17(3):167–180.
- Collemare J, Billard A, Böhnert HU, et al. Biosynthesis of secondary metabolites in the rice blast fungus Magnaporthe grisea: the role of hybrid PKS-NRPS in pathogenicity. Mycol Res. 2008;112:207–215.
- Cimermancic P, Medema MH, Claesen J, et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell. 2014;158:412–421.
- Clevenger KD, Bok JW, Ye R, et al. A scalable platform to identify fungal secondary metabolites and their gene clusters. Nat Chem Biol. 2017;13:895–901.
- Nielsen JC, Grijseels S, Prigent S, et al. Global analysis of biosynthetic gene clusters reveals vast potential of secondary metabolite production in Penicillium species. Nat Microbiol. 2017;2:17044.
- Hyatt D, Chen GL, Locascio PF, et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119.
- Aherfi S, Andreani J, Baptiste E, et al. A large open pangenome and a small core genome for giant pandoraviruses. Front Microbiol. 2018;9:1486.
- Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189.
- Schoch CL, Seifert KA, Huhndorf S, et al.; Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA. 2012;109:6241–6246.
- Blaalid R, Kumar S, Nilsson RH, et al. ITS1 versus ITS2 as DNA metabarcodes for fungi. Mol Ecol Resour. 2013;13:218–224.
- Bengtsson‐Palme J, Ryberg M, Hartmann M, et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol Evol. 2013;4:914–919.
- Lesage-Meessen L, Haon M, Uzan E, et al. Phylogeographic relationships in the polypore fungus Pycnoporus inferred from molecular data. FEMS Microbiol Lett. 2011;325:37–48.
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797.
- Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–577.
- Retief JD. Phylogenetic analysis using PHYLIP. Methods Mol Biol. 1999;132:243–258.
- Tamura K, Peterson D, Peterson N, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739.
- Tatusov RL, Fedorova ND, Jackson JD, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41.
- Huerta-Cepas J, Szklarczyk D, Forslund K, et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016;44:D286–D293.
- Lombard V, Golaconda Ramulu H, Drula E, et al. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D495.
- Cantarel BL, Coutinho PM, Rancurel C, et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37:D233–D238.
- Mäkelä M, DiFalco M, McDonnell E, et al. Genomic and exoproteomic diversity in plant biomass degradation approaches among Aspergilli. Stud Mycol. 2018;91:79–99.
- Lomascolo A, Cayol JL, Roche M, et al. Molecular clustering of Pycnoporus strains from various geographic origins and isolation of monokaryotic strains for laccase hyperproduction. Mycol Res. 2002;106:1193–1203.
- Levasseur A, Lomascolo A, Chabrol O, et al. The genome of the white-rot fungus Pycnoporus cinnabarinus: a basidiomycete model with a versatile arsenal for lignocellulosic biomass breakdown. BMC Genomics. 2014;15:486.
- Busk PK, Lange M, Pilgaard B, et al. Several genes encoding enzymes with the same activity are necessary for aerobic fungal degradation of cellulose in nature. PLoS One. 2014;9:e114138.
- Couturier M, Navarro D, Chevret D, et al. Enhanced degradation of softwood versus hardwood by the white-rot fungus Pycnoporus coccineus. Biotechnol Biofuels. 2015;8:216.
- Pavlov AR, Tyazhelova TV, Moiseenko KV, et al. Draft genome sequence of the fungus Trametes hirsuta 072. Genome Announc. 2015;3:e01287–01215.
- Cerrón LM, Romero-Suárez D, Vera N, et al. Decolorization of textile reactive dyes and effluents by biofilms of Trametes polyzona LMB-TM5 and Ceriporia sp. LMB-TM1 isolated from the Peruvian Rainforest. Water Air Soil Pollut. 2015;226:235.
- Granchi Z, Peng M, Chi-A-Woeng T, et al. Genome sequence of the basidiomycete white-rot fungus Trametes pubescens FBCC735. Genome Announc. 2017;5:e0164301616.
- Wang J, Zhang Y, Xu Y, et al. Genome sequence of a laccase producing fungus Trametes sp. AH28-2. J Biotechnol. 2015;216:167–168.
- Floudas D, Binder M, Riley R, et al. The Paleozoic origin of enzymatic mechanisms for decay of lignin reconstructed using 31 fungal genomes. Science. 2012;336:1715–1719.
- de Oliveira Carneiro RT, Lopes MA, Silva MLC, et al. Trametes villosa lignin peroxidase (TvLiP): genetic and molecular characterization. J Microbiol Biotechnol. 2017;27:179–188.
- Rytioja J, Hildén K, Yuzon J, et al. Plant-polysaccharide-degrading enzymes from basidiomycetes. Microbiol Mol Biol Rev. 2014;78:614–649.
- Berrin JG, Navarro D, Couturier M, et al. Exploring the natural fungal biodiversity of tropical and temperate forests toward improvement of biomass conversion. Appl Environ Microbiol. 2012;78:6483–6490.
- Yin Y, Mao X, Yang J, et al. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40:W445–W451.
- Murphy C, Powlowski J, Wu M, et al. Curation of characterized glycoside hydrolases of fungal origin. Database (Oxford). 2011;2011:bar020.
- Viborg AH, Terrapon N, Lombard V, et al. A subfamily roadmap of the evolutionarily diverse glycoside hydrolase family 16 (GH16). J Biol Chem. 2019;294:15973–15986.