
Abstract
An endolichenic fungus, Xylaria grammica strain EL000614, showed strong nematicidal effects against plant pathogenic nematode, Meloidogyne incognita by producing grammicin. We report genome assembly of X. grammica EL000614 comprised of 25 scaffolds with a total length of 54.73 Mb, N50 of 4.60 Mb, and 99.8% of BUSCO completeness. GC contents of this genome were 44.02%. Gene families associated with biosynthesis of secondary metabolites or regulatory proteins were identified out of 13,730 gene models predicted.
Lichens provide habitats for many microorganisms, including endolichenic fungi. The endolichenic fungi have tremendous potential to produce bioactive metabolites. Compounds produced by endolichenic fungi have been shown to exhibit antitumor, antibacterial, antifungal, and nematicidal activities [Citation1,Citation2]. One endolichenic fungus, Xylaria grammica EL000614, showed strong nematicidal activity against deleterious plant pathogenic nematode, Meloidogyne incognita, causing root-knot disease on a wide range of host plants [Citation2]. The active compound was purified and identified as grammicin [Citation2]. Grammicin is an isomer of patulin, a notorious mycotoxin showing antibacterial and cytotoxic activity [Citation3]. Unlike patulin, however, grammicin did not show antibacterial activities against various plant pathogenic bacteria, nor cytotoxic activity toward the human first-trimester trophoblast cell line SW.71 [Citation2]. As a first step to reveal the grammicin biosynthetic pathway, the genome of EL000614 was sequenced.
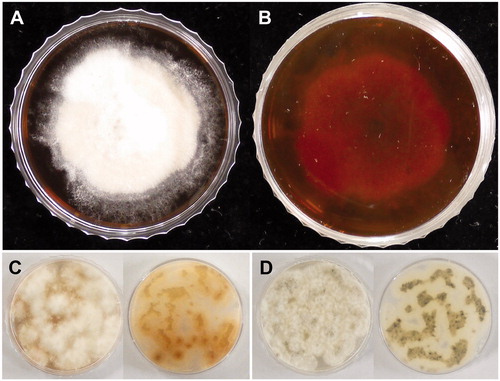
Colonies of X. grammica strain EL000614 were yellowish-white on the front () and dark red on the reverse side () when grown for two weeks on potato dextrose agar medium. This dark red pigment was accumulated only in the dark (), but not under constant light condition (). Black pigment was accumulated in illuminated plates. The fungus was grown in potato dextrose broth for 2 days at 23 °C, and genomic DNA was extracted from young mycelium using the DNeasy mini kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions.
Figure 1. Morphological characteristics of Xylara grammica strain EL000614. (A) Two weeks-old colony on the upper side of PDA. (B) Dark red pigmentation on the reverse side of PDA. Four-day-old colony on the upper side (left) and the reverse side (right) of PDA under constant dark condition (C) and under constant light condition (D).
Whole-genome sequencing was performed by the Theragen Etex Bio Institute (Suwon, Korea) using a combination of Hiseq 2000 (IIIumina, CA, USA) and PacBio RS II (Pacific Biosciences, CA, USA). A short-insert (fragment size of 400 bp) paired-end (PE) library was constructed using the TruSeq DNA sample prep kit (Illumina), and two long-insert mate-pair (MP) libraries (insert sizes of 5 kb and 10 kb, respectively) were generated using a Nextera mate-pair library prep kit (Illumina). Eight PacBio RS II single-molecule real-time (SMRT) cells (total of 943,964 reads with an average read size of 7,891 bp) were obtained according to the PacBio standard library preparation protocol using the PacBio DNA Template Prep kit with the average fragment size of 20 kb. The genome was de novo assembled using the overlap-layout-consensus (OLC) algorithm in CANU v1.8 [Citation4,Citation5]. The assembled genome of long-reads was polished based on Illumina paired-end library (70,717,638 reads) using Pilon v 1.22 [Citation6]. The final genome consists of 25 contigs and one mitochondrial genome with a total length of 54,730,220 bp and 196,363 bp, respectively. The assembly has N50 being 4.60 Mbp, and the GC content of the genome was 44.02%. Genome completeness was assessed using BUSCO v. 5.0.0 against the “fungi_odb10” database, showing 99.8% of 758 fungal gene sets were complete () [Citation7].
Table 1. Genome assembly and annotation statistics of Xylaria grammica EL000614.
Gene prediction was performed by BRAKER2 [Citation8] using constructed model from RNA-seq and protein models of five close species in the NCBI nonredundant (nr) database. A total of 13,730 gene models were predicted, of which 12,900 genes (93.96%) had hits in UniProt, nr, or InterPro database. Ninety-eight Biosynthetic gene clusters (BGCs), including 45 polyketide synthase (PKS) and 42 non-ribosomal protein synthetase (NRPS), 21 terpene synthase, were identified through antiSMASH v5.0 [Citation9]. Among these were two orthologs encoding PatK, a PKS backbone gene in patulin biosynthesis [Citation10]. Further, 476 genes encoding transcription factor and 222 genes encoding cytochrome P450 were cataloged through Fungal Transcription Factor Database (FTFD) v1.0 [Citation11], and Fungal Cytochrome P450 Database v1.0 [Citation12], respectively. Genome was composed of 16.36% of repetitive sequences such as retrotransposons, DNA transposons, simple sequence repeats, and unclassified repeats through the combining of RebBase [Citation13,Citation14] search and de novo identification using RepeatModeler 1.0.8 and RepeatMasker 4.0.6 [Citation15]. A total of 531 non-coding RNAs, including 43 rRNA from BLASTn search against NCBI, 225 tRNA through tRNAscan-SE [Citation16], 121 microRNA and 142 small nucleolar RNA through Infernal search against Rfam 11.0 [Citation17], were also identified. This draft genome of X. grammica EL000614 will help further characterize the genes involved in grammicin biosynthesis and their regulation.
The sequence data and RNA-seq data of X. grammica EL000614 were deposited at Sequence Read Archive (SRA) under the accession number SRR14073447 to SRR14073457. The assembly data also deposited GenBank under the accession no. NGZP00000000. The version described in this article is NGZP02000000 (https://www.ncbi.nlm.nih.gov/nuccore/NGZP00000000.2). The associated Bioproject number is PRJNA368785
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Kellogg J, Raja HA. Endolichenic fungi: a new source of rich bioactive secondary metabolites on the horizon. Phytochem Rev. 2017;16(2):271–293.
- Kim TY, Jang JY, Yu NH, et al. Nematicidal activity of grammicin produced by Xylaria grammica KCTC 13121BP against Meloidogyne incognita. Pest Manag Sci. 2018;74(2):384–391.
- Edwards RL, Maitland DJ, Pittayakhajonwut P, et al. Metabolites of the higher fungi. Part 33. Grammicin, a novel bicyclic C7H6O4 furanopyranol from the fungus Xylaria grammica (Mont.) Fr. Perkin Trans. 2001;1(11):1296–1299.
- Wang A, Wang Z, Li Z, et al. BAUM: improving genome assembly by adaptive unique mapping and local overlap-layout-consensus approach. Bioinformatics. 2018;34(12):2019–2028.
- Koren S, Walenz BP, Berlin K, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27(5):722–736.
- Walker BJ, Abeel T, Shea T, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. 2014;9(11):e112963.
- Waterhouse RM, Seppey M, Simao FA, et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 2018;35(3):543–548.
- Brůna T, Hoff KJ, Lomsadze A, et al. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP + and AUGUSTUS supported by a protein database. NAR Genom Bioinform. 2021;3:108.
- Blin K, Shaw S, Steinke K, et al. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019;47(W1):W81–W87.
- Artigot MP, Loiseau N, Laffitte J, et al. Molecular cloning and functional characterization of two CYP619 cytochrome P450s involved in biosynthesis of patulin in Aspergillus clavatus. Microbiology. 2009;155(Pt 5):1738–1747.
- Park J, Park J, Jang S, et al. FTFD: an informatics pipeline supporting phylogenomic analysis of fungal transcription factors. Bioinformatics. 2008;24(7):1024–1025.
- Park J, Lee S, Choi J, et al. Fungal cytochrome P450 database. BMC Genomics. 2008;9:402.
- Bao W, Kojima KK, Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob Dna. 2015;6:11.
- Kapitonov VV, Jurka J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat Rev Genet. 2008;9(5):411–412.
- Smit AFA, Hubley R, Green P. RepeatMasker. 2015. Available from: http://repeatmasker.org.
- Chan PP, Lowe TM. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol. 2019;1962:1–14.
- Burge SW, Daub J, Eberhardt R, et al. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013;41(Database issue):D226–D232.