
Abstract
Fungal endophytes have been recorded in various plant species with a richness of diversity, and their presence plays an essential role in host plant protection against biotic and abiotic stresses. This study applied the Illumina MiSeq sequencing platform based on the amplification of fungal ribosomal ITS2 region to analyze fungal endophytic communities of two oak species (Quercus mongolica and Q. serrata) with different oak wilt disease susceptibilities in Korea. The results showed a total of 230,768 sequencing reads were obtained and clustered at a 97% similarity threshold into 709 operational taxonomic units (OTUs). The OTUs of Q. serrata were higher than that of Q. mongolica with the number of 617 OTUs and 512 OTUs, respectively. Shannon index also showed that Q. serrata had a significantly higher level of fungal diversity than Q. mongolica. Total of OTUs were assigned into 5 fungal phyla, 17 classes, 60 orders, 133 families, 195 genera, and 280 species. Ascomycota was the dominant phylum with 75.11% relative abundance, followed by Basidiomycota with 5.28%. Leptosillia, Aureobasidium and Acanthostigma were the most abundant genera detected in Q. serrata with the average relative abundance of 2.85, 2.76, and 2.19%, respectively. On the other hand, Peltaster, Cladosporium and Monochaetia were the most common genera detected in Q. mongolica with the average relative abundance of 4.83, 3.03, and 2.87%, respectively. Our results indicated that fungal endophytic communities were significantly different between two oak species and these differences could influence responses of host trees to oak wilt disease caused by Raffaelea quercus-mongolicae.
1. Introduction
Endophytes are microorganisms that colonize internal living tissues of plant, but without causing any immediate over adverse effects or during a part of their life cycle reside inside plant tissue of host trees without doing substantive harm [Citation1,Citation2]. Endophytes may include bacterial and fungal species or others belong to actinomycetes and mycoplasma [Citation1]. In addition, bioactive compounds produced by endophytes play an important role in fitness enhancements for host plants [Citation1,Citation3]. On the other hand, endophytes can improve the ability of plants' nutrient uptake and protect host plants against biotic and abiotic stresses [Citation3,Citation4]. Fungal endophytes have been used to extract antimicrobial compounds and produce antibiotic drugs [Citation1]. Endophytic fungi have been recorded in various plant species with a richness of diversity [Citation1,Citation2]. The most common endophytic fungi that have been identified belong to Ascomycota, followed by Basidiomycota and other fungi [Citation1,Citation2].
It is well known that metagenomics based on next-generation sequencing (NGS) technologies helps scientists to discover and understand the diversity, function, and evolution of the uncultivated microbiology of diverse environments or habitats [Citation5]. Metagenomics is considered as an optimal tool to discover the nucleic acids from uncultivated microbes in different environments [Citation5,Citation6]. Metagenomics is defined as a technical chain rooted in genomics, microbial genetics, microbial ecology, and bioinformatics to analyze directly genomes of microbial community sampled from their natural habitats [Citation5]. It is also applied to recover target genes through sequencing to determine not only the microbial diversity but also their roles in environmental samples [Citation5–7]. Among the NGS technologies, Pyrosequencing (Roche 454) and Solexa (Illumina) sequencing systems have been extensively used to explore microbial communities in different environments [Citation5]. For instance, results of 454 sequencing indicated that fungal communities in the phyllosphere of Quercus macrocarpa were significantly different either between urban and nonurban environments or between different seasons [Citation8,Citation9]. Differences in bacterial and fungal endophytic communities between Acer campester and A. platanoides leaves were deciphered by using Illumina MiSeq sequencing [Citation10]. Fungal endophytic communities in needles of Pinus sylvestris were also analyzed by using Illumina MiSeq sequencing, and the study showed the most abundant fungi was assigned to phylum Ascomycota with 91.2% of the samples [Citation11]. In addition to the two above-mentioned, the Ion Torrent Personal Genome Machine (PGM) sequencing was also applied to analyze endophytic fungal communities in Eucalyptus grandis [Citation12] or investigate microbial community dynamic in liquid waste [Citation13].
Quite a few studies about endophytic fungi in Korea have been studied in recent years [Citation14–19]. For instance, Alternaria spp., Cladosporium spp., and Penecillium spp. were the dominant endophytic fungi in several medicinal plants [Citation14]. Species richness and diversity indices of endophytic fungi were different among three Halophytes, namely Sedum oryzifolium, Lysimachia mauritiana, and Aster spathulifolius [Citation17]. The most common genera of endophytic fungi isolated from Pinus thunbergii were identified as Fusarium, Penicillium, and Trichoderma [Citation16], while Lophodermium conigenum and Annulohypoxylon turcatum were dominant in Pinus densiflora and Juniperus rigida, respectively [Citation18]. However, endophytic fungi obtained from P. densiflora and J. rigida were influenced by host plants’ distribution and had a lower diversity index than that of Larix kaempferi [Citation15]. Studies on endophytic fungi of other coniferous trees, namely Cryptomeria japonica, Pinus koraiensis, Pinus rigida, etc. have been also reported, with a total of about 80 taxa belonged to 52 genera [Citation19].
Applying metagenomics to analyze microbial community from various environments was also conducted in Korea [Citation20–26]. Of which, NGS platform of Roche 454 was applied to identify airborne fungal community in Seoul [Citation21], while Illumina MiSeq platform was performed to compare soil higher fungal communities associated with dead and living Korean fir (Abies koreana) in Jeju island [Citation26]. However, using metagenomics to analyze endophytic fungal communities in forest trees is still limited. Quercus serrata was known as the most susceptible oak species to the oak wilt fungus Raffaelea quercivora in Japan [Citation27], but Quercus mongolica is highly susceptible to the oak wilt fungus Raffaelea quercus-mongolicae, and Q. serrata is relatively resistant to this pathogen in Korea [Citation28]. The difference in susceptibility of these two oak species to R. quercus-mongolicae could be partially due to differences in their endophytic fungal communities. Hence, our objectives in the present study were to (i) compare species richness and diversity of endophytic fungi between Q. mongolica and Q. serrata and to (ii) characterize taxonomic structures of endophytic fungi that differ between two oak species through Illumina MiSeq sequencing platform.
2. Materials and methods
2.1. Sampling collections
Samples were collected in triplicate from different organs of healthy trees (leaf, petiole, twig, branch, stem, and root) of two oak species (Q. mongolica and Q. serrata) in Chuncheon city, Gangwon province, Korea (37°52’12” N, 127°45’58” E) in September 2019. Leaf, petiole, twig, branch, and root samples were collected by pruning shears, while stem samples were collected by increment borer at breast height position on the trees. All samples were then kept in plastic bags with coolers to bring to the laboratory. The root samples were gently washed with tap water to remove soil. Surface disinfection of samples was performed with solutions including 2.0% NaOCl for 3 min, followed by 70% ethanol for 1 min. Then, samples were washed with sterilized water for 15 sec in three times. Finally, all samples were dried using sterilized tissue paper, cut in 0.5 × 0.5 cm size (leaf), with 0.5 cm length (other tissues), and stored at −86 °C until DNA extraction.
2.2. DNA extraction
Samples were shredded and then grinded using Tube Mill Control (IKA-Werke GmbH & CO. KG, Staufen, Germany) with speed 5000 rpm for 3 min. The DNA was purified by using Quick-DNA Fungal/Bacterial Miniprep Kit (Zymo Research, Irvine, CA, USA), following instructions of the manufacturer. The final 20 µl pure DNA eluted from each sample. To prepare for amplification, pure DNA from each sample of three replications was pooled and a total of 12 DNA samples (6 samples for each oak species) was stored at −20 °C until amplification.
2.3. ITS amplification and DNA library construction
The quantification and quality of extracted DNA were determined using Quant-IT PicoGreen (Invitrogen, Molecular Probes, Eugene, OR, USA). After performing quality control (QC), qualified samples proceed to library construction. The sequencing libraries are prepared according to the Illumina fungal metagenomic sequencing demonstrated protocol to amplify the fungal ribosomal internal transcribed spacer second (ITS2) region [Citation29]. The universal primer pair with Illumina overhang adapter sequences used for amplicon PCR were as follows: ITS-3F (5′-GCATCGATGAAGAACGCAGC-3′) and ITS-4R (5′-TCCTCCGCTTATTGATATGC-3′). The input gDNA of each sample (5 ng/µl) was amplified with 2x KAPA HiFi HotStart ReadyMix (Kapa Biosystems, Inc., Wilmington, MA, USA) and 500 nM each of the universal forward/reverse PCR primer. The cycle conditions for the first PCR were 3 min at 95 °C for heat activation, and 25 cycles of denaturation for 30 sec at 95 °C, annealing for 30 sec at 55 °C and extension for 30 sec at 72 °C, followed by a 5 min final extension at 72 °C. The first PCR products were analyzed on Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) using Agilent DNA 1000 Kit (Agilent Technologies, Waldbronn, Germany) and purified then with the AMPure XP beads (Beckman Coulter, Indianapolis, IN, USA). The procedure of analysis and cleanup for the first PCR products were conducted following the manufacturer's instructions. The 5 µl of the first PCR product was used in dual-indexed PCR amplification for final library construction using the Nextera XT Index Kit v2 (Illumina, San Diego, CA, USA). The cycle conditions for the second PCR were same as the first PCR conditions, but only 8 cycles were performed. The AMPure XP beads (Beckman Coulter) were used to clean up the final library before quantification. The library was quantified using a Bioanalyzer DNA 1000 chip (Agilent Technologies, Waldbronn, Germany). The amplicon libraries were then normalized and pooled at equimolar concentrations before sequencing. The paired-end (v3, 2 × 300 bp) sequencing was performed by the Macrogen Inc. using the Illumina MiSeq platform (Seoul, Korea).
2.4. Sequencing processing and taxonomical assignment
Reads from paired-end sequences were merged using FLASH (Fast Length Adjustment of SHort reads) program [Citation30] with default parameters. Pre-processing and clustering were conducted using CD-HIT-OTU-MiSeq program [Citation31,Citation32] with steps as follows: short reads were filtered out and trimmed to remove bases with a Phred quality score less than 20; filtered reads were then clustered at 100% identify using CD-HIT-DUP to remove duplicates and identify chimeric reads; secondary clusters were recruited into primary clusters; chimeric reads and small clusters with below 140 bp were removed. The remaining reads from nonchimeric clusters were clustered using a greedy algorithm into Operational Taxonomic Unit (OTUs) at a 97% similarity threshold. Representative sequences from each OTU were used to assign taxonomy using UCLUST classifier in QIIME [Citation33] against the UNITE database [Citation34]. Unclassified OTUs and nonfungal OTUs were removed using the UNITE + INSD Fungi Version 8.2 (Feb 2020) to generate only fungal OTU table and taxonomy.
2.5. Statistical analysis and computations
Alpha-diversity indices of fungal community such as observed OTUs, Chao1, Shannon index, and so on were calculated using the alpha_diversity.py and alpha_rarefaction.py, while the beta-diversity distance matrix was computed based on UniFrac analysis, and weighted UniFrac distances were used to construct principal coordinates analysis (PCoA) plots using make_2d_plots.py workflow in QIIME to visualize similarities in the fungal community composition. Differences in alpha diversity between two oak species were compared by the independent samples t-test at 5% probability level using R program [Citation35]. The relative abundances of OTU proportion for data from different taxonomical levels (phyla, class, and genera) were also graphed using R program [Citation35].
3. Results
3.1. Fungal diversity and richness
A De Novo Genome Assembly resulted that the total bases obtained from Illumina sequencer are 544,250,767 bp and 1,424,349 raw tags without ambiguous base (). The GC content percentage ranged from 54.54 to 61.53% and the percentages of bases with Q20 were more than 99.0%, while the rates of bases with Q30 were above 95.0% (). These results demonstrated that the data set from Illumina sequencer had good quality.
Table 1. Summary of genome assembly of two Quercus species by FLASH program per sample.
The results of preprocessing obtained in a total number of 230,768 sequence reads (clean tags) with a minimal and maximal number of 2885 and 49,059 reads, respectively (). These sequences were clustered into 709 OTUs (ranging from 92 to 390) at a 97% similarity level ( and ). The sequencing depth and species richness was described using rarefaction analysis in the . The total OTUs of Q. serrata were higher than that of Q. mongolica with the total number of 617 OTUs and 512 OTUs, respectively (); however, the average of OTUs was not significantly different (p > 0.05) between two oak species ( and ). Although two oak species had no significant differences (p > 0.05) in the average Chao1 richness, samples from each plant tissue of Q. serrata had relatively higher Chao1 richness than those of Q. mongolica ( and ). Shannon index also showed that Q. serrata had a significantly higher level (p < 0.05) of fungal diversity than Q. mongolica, and the petiole sample of Q. serrata was the most diverse with Shannon index of 5.76, while the lowest diversity with Shannon index of 2.65 belonged to stem sample of Q. mongolica ( and ). Inverse Simpson index also indicated that Q. serrata had a higher level of fungal diversity than Q. mongolica, however, this difference was not significant (p > 0.05) between two oak species ( and ). Good's coverage index gives a relative measure of how well the sample represents the larger environment. In our study, Good's coverage index of twelve samples was between 98.34 and 99.89% ().
Figure 1. Venn diagram showing the number of fungal OTUs overlapped and nonoverlapped from Quercus mongolica and Quercus serrata.
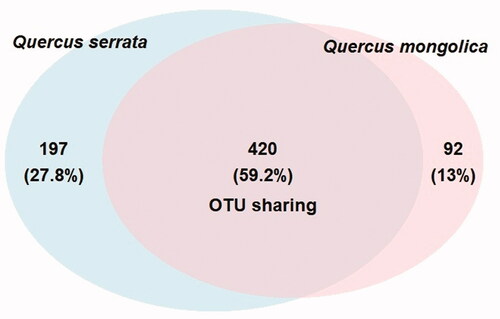
Figure 2. Rarefaction curves described for observed OTUs metric (left) and chao1 metric (right) among all 12 samples (QS: Quercus serrata; QM: Quercus mongolica).
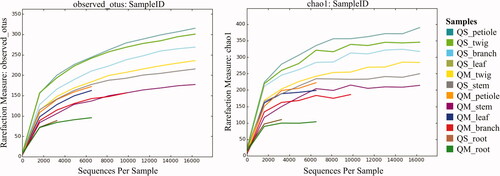
Figure 3. Fungal community richness (A) and diversity; (B) in Quercus mongolica and Quercus serrata described by boxplot. *Indicate a significant difference (p < 0.05) by two sample t-test.
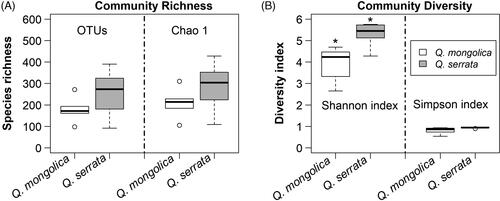
Table 2. Summary of community richness and alpha diversity per sample.
3.2. Taxonomic assignment of endophytic fungi
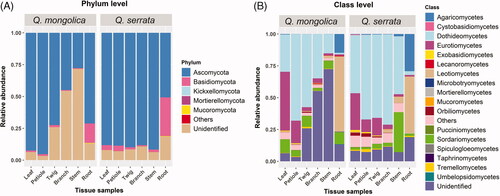
Total of OTUs was assigned into five fungal phyla, 17 classes, 60 orders, 133 families, 195 genera, and 280 species (Supplementary Table S1–S5). Fungal endophytes colonized in Q. mongolica belonged to five fungal phyla, 16 classes, 52 orders, 110 families, 157 genera, and 220 species, while those of Q. serrata included four fungal phyla, 17 classes, 59 orders, 127 families, 180 genera, and 246 species (Supplementary Table S1-S5). Members of Ascomycota were the most common endophytic fungi among all samples with 75.11%, followed by unidentified fungi with 19.50% and Basidiomycota with 5.28%, while Mucoromycota, Mortierellomycota, Kickxellomycota and others were found with very low rate of 0.07, 0.02, 0.01, and 0.01%, respectively (). The percentage of Ascomycota ranged from 44.88 to 95.28% among samples of Q. mongolica, whereas that of Q. serrata was between 50.68 and 91.65% (). Basidiomycota accounted for 0.63 to 15.15% and 0.88 to 30.22% in Q. mongolica and Q. serrata respectively (). Mucoromycota and Mortierellomycota were mainly detected in root and stem samples in both two oak species; however, Kickxellomycota was only detected in root sample of Q. mongolica ( and Supplementary Table S1). Members of fungal classes also varied in different samples, in which, Dothideomycetes, Eurotiomycetes, Leotiomycetes and Sordariomycetes were dominant within Ascomycota with average relative abundances of 41.91, 11.01, 9.70 and 6.94%, respectively ( and Supplementary Table S1). Agaricomycetes had the highest proportions within Basidiomycota and this fungal class mainly distributed in root samples of Q. mongolica and Q. serrata with relative abundances of 14.78 and 29.20% respectively ( and Supplementary Table S1).
Figure 4. Relative abundance of endophytic fungi at the phylum (A) and class level; (B) in 12 different tissue samples between Quercus mongolica and Quercus serrata.
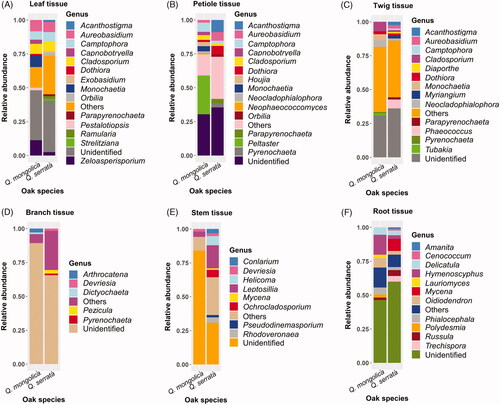
There were 9 and 10 detected genera with average relative abundance of more than 1% in Q. mongolica and Q. serrata, respectively. In which, three genera of Aureobasidium, Camptophora and Cladosporium were distributed in both two oak species (Supplementary Table S1). Leptosillia, Aureobasidium and Acanthostigma were the most abundant genera detected in Q. serrata with the average relative abundance of 2.85, 2.76 and 2.19%, respectively. On the other hand, Peltaster, Cladosporium and Monochaetia were the most common genera detected in Q. mongolica with the average relative abundance of 4.83, 3.03 and 2.87%, respectively (Supplementary Table S1).
Relative abundance of different fungal genera in various plant tissues between two oak species was described in the . The number of fungal genera with relative abundances above 1.0% in leaf, petiole, and twig tissue was higher than that in branch, stem, and root tissue (). The most abundant genera in leaf, petiole, and twig of Q. mongolica and Q. serrata were Zeloasperisporium (11.29%) and Aureobasidium (7.35%), Peltaster (28.5%) and Acanthostigma (8.87%), Cladosporium (7.01%) and Phaeococcus (6.57%), respectively (). The most common genera presented in both branches and stem of Q. mongolica were Arthrocatena (2.81%) and Leptosillia (3.82%), respectively, while Pezicula (2.62%) and Leptosillia (17.04%) were the most abundant genera in branch and stem of Q. serrata, respectively (). The root tissue of both Q. mongolica and Q. serrata had a difference in the diversity of fungal genera compared to other tissues and this difference was due to the presence of several genera belong to Basidiomycota such as Delicatula (5.51%), Russula (1.84%), Mycena (8.93%), and so on ( and Citation6). Hymenoscyphus (14.66%) was the most abundant genus in root of Q. mongolica, while Mycena (8.93%) accounted for the highest relative abundance in root of Q. serrata ().
Figure 5. Relative abundance of different fungal genera in various plant tissues between Quercus mongolica and Quercus serrata. The fungi with a relative abundance below 1% were grouped into others.
4. Discussion
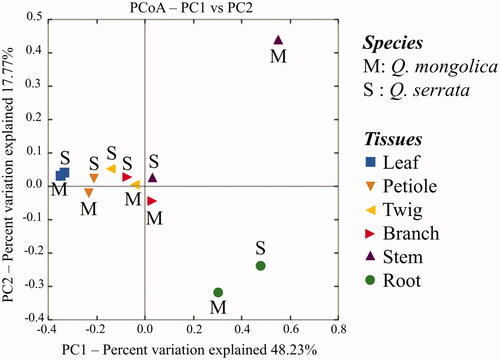
A total of 709 distinct OTUs were generated from two oak species (). Among these, there were 59.2% OTUs were distributed in both Q. serrata and Q. mongolica, while 27.8 and 13.0% OTUs were only detected in Q. serrata and Q. mongolica, respectively (). It was indicated that Q. serrata is highly resistant to oak wilt fungus (R. quercus-mongolicae), while Q. mongolica is highly susceptible to this pathogen [Citation28]. Our results showed that the Shanon index of endophytic fungi in Q. serrata was significantly higher than that of Q. mongolica (). This is consistent with previous findings that either endophytic fungi of disease-resistant species had a higher diversity index compared with susceptible species to disease [Citation36] or fungal endophytic community had a significant difference between pathogen-infected trees and healthy trees [Citation10,Citation37,Citation38]. For instance, endophytic fungi colonized in Rosa multiflora had higher diversity than Rose multiflora var. carnea in several specific developmental stages of plants where R. multiflora showed high resistance to powdery mildew disease and R. multiflora var. carnea was highly susceptible to this pathogen [Citation36]. Two maple trees, Acer campestre and Acer platanoides had a significantly higher fungal richness and diversity in control leaves compared to leaves infected with pathogens [Citation10], while European beech (Fagus sylvatica) showed a significant difference in the fungal species composition between living and decaying leaves [Citation38]. Fungal diversity indices were different among plant tissues of two oak species (). Principal Coordinate Analysis (PCoA) also showed that the fungal endophytic community had a difference in various plant tissues (leaf, petiole, twig, branch, stem, and root), and the biggest difference between two oak species was indicated in stem samples (). Fungal endophytic communities were affected not only by plant varieties, plant tissues but also by management practices, plant's development stages, and hosts' ecological conditions [Citation8,Citation9,Citation36,Citation39]. The diversity of endophytic fungi isolated from plants was also different among parts [Citation40–42] with a higher abundance in leaves compared to other tissues [Citation40,Citation41]. Our study obtained that the highest diversity index of endophytic fungi was indicated in the leaf sample of Q. mongolica, while endophytic fungi of Q. serrata showed the highest diversity index in the petiole sample ().
Figure 6. Principal Coordinate Analysis (PCoA) based on the weighted UniFrac distances for fungal communities from plant tissues of two oak species (Quercus mongolica and Quercus serrata) indicating two distinct groups in the stem tissues between two oak species using two principal coordinates (PC1 and PC2).
Ascomycota was dominant in both Q. mongolica and Q. serrata, with the average relative abundances of 67.34 and 82.88%, respectively ( and Supplementary Table S1). Ascomycota is the most dominated endophytic fungi colonized in many plants that have been indicated in previous studies [Citation11,Citation36,Citation39,Citation43–45]. The most abundant fungal genus in Q. mongolica was Peltaster with average relative abundances of 4.83% and this genus was mainly colonized in petiole tissue with relative abundances of 28.50%, while Leptosillia was the most common endophytic fungi in Q. serrata with average relative abundances of 2.85% and it was mainly colonized in stem tissue with relative abundances of 17.04% ( and Supplementary Table S1). To date, the information about endophytic fungi identified as Peltaster spp. and Leptosillia spp. is still limited.
Cladosporium spp. and Aureobasidium spp. were dominant endophytic fungi in both Q. mongolica and Q. serrata (Supplementary Table S1). Cladosporium spp. accounted for 3.03 and 1.99% of endophytic fungi in Q. mongolica and Q. serrata respectively, while Aureobasidium spp. had 1.80% in Q. mongolica and 2.76% in Q. serrata (Supplementary Table S1). Cladosporium spp. have been reported as dominant fungal endophyte from various either host trees such as oak, ash, and cinnamon species [Citation46–48] or other hots plants, such as sweet citrus [Citation49], rice [Citation50], and wheat [Citation51]. Aureobasidium spp. was one of the most common endophytic fungi colonized in several tree species [Citation52], of which, endophytic fungus Aureobasidium pullulans accounted for 24.2% of detected fungi in European ash (Fraxinus excelsior) [Citation53] and was the most dominant taxon in European aspen (Populus tremula) [Citation54]. Moreover, Aureobasidium pullulans was also one of major endophytic fungi in maize and common bean [Citation55,Citation56]. Studies on bioactive compounds obtained from Cladosporium spp. and Aureobasidium spp. were also conducted in biological controls of plant pathogens [Citation57–59]. For instance, Cladosporium spp. isolated from stem of Sesbania grandiflora can inhibit development of bacterial and fungal pathogens in Thailand such as Staphylococcus aureus, Escherichia coli, and Cryptococcus neoformans [Citation57]; Cladosporium sp., an endophytic fungus isolated from a medicinal plant (Cyclosorus parasiticus), had antibacterial activity against Staphylococcus aureus and Salmonella enterica damaging on C. parasiticus [Citation60]. In addition, extracellular enzymatic activities of Cladosporium spp. isolated from various medicinal plants were also reported [Citation61,Citation62]. On the other hand, Aureobasidium spp. isolated from Posidonia oceanica can produce antimicrobial compounds, namely hydroxylated decanoic acids against Candida albicans and Staphylococcus aureus, and aureobasidin which exhibited insecticidal activity against the larval settlement of Balanus amphitrite larvae [Citation59]. Among Aureobasidium spp., A. pullulans has been identified as one of the biocontrol agents to control various fruit postharvest pathogens such as Monilinia laxa on plums, peaches, sweet cherries, apricots, and table grapes [Citation63–66]; Botrytis cinerea on apples, sweet cherries, tomatoes. and table grapes [Citation63,Citation66–68]; Penicillium expansum on apples, lemons [Citation63,Citation67,Citation69]; Monilinia fructicola, Monilinia polystroma and Monilinia fructigena on sweet cherries, peaches, and apricots [Citation64,Citation65]; Colletotrichum acutatum on apples [Citation67]; Penicillium italicum and Penicillium digiatum on citruses, apples, and lemons [Citation67,Citation69]. Aureobasidium pullulans was also used to control plant pathogens, namely Phytophthora infestans causing tomato late blight [Citation70], Rhizoctonia solani causing damping-off in tomato, bean, and soybean seedlings [Citation71,Citation72], Fusarium culmorum causing Fusarium head blight of common wheat (Triticum aestivum) [Citation73], and Neofusicoccum parvum causing stem canker disease in apple trees [Citation74]. Other phytopathogenic fungi such as Fusarium oxysporum and Alternaria alternata were also inhibited by A. pullulans isolated from healthy grapevines [Citation75]. Moreover, A. pullulans was also identified as plant growth promoters for bean and soybean plants [Citation72], while Aureobasidium sp. isolated from Boswellia sacra not only displayed extracellular enzymatic activities but also produced indole acetic acid (IAA) for promoting growth of B. sacra [Citation76]. In brief, Aureobasidium spp. had a higher abundance in Q. serrata than Q. mongolica, and these fungi showed a wide range of bioactive activities, especially antifungal and insecticidal activities. Thus, they could play important roles in pathogen tolerance and repellent of insect vector in oak wilt pathosystem.
Oak wilt disease caused by R. quercus-mongolicae has emerged rapidly in Korea since 2004 [28], and Ceratocystis quercicola, a novel Ceratocystis species can cause a very low level of damage on Quercus variabilis [Citation77]. However, these two fungal species were not found in the present study. To the best of our knowledge, this is the first study that was conducted to analyze endophytic fungal community from Q. mongolica and Q. serrata based on the ITS2 region through Illumina MiSeq. Our results provided a better understanding of differences in diversity of fungal endophyte colonized in these two oak species. These differences could affect the interactions between endophytic fungi and host tree species in producing specific enzymes or volumes of bioactive compounds, therefore, the responses of Q. mongolica and Q. serrata to oak wilt pathogen are different.
5. Conclusion
Our results indicate that the diversity of endophytic fungi was significantly different between two oak species, and the biggest difference of fungal endophytic community occurred in stem tissues. A total of 709 OTUs were obtained in both two oak species. In which, the OTUs of Q. serrata were higher than that of Q. mongolica with the number of 617 OTUs and 512 OTUs, respectively. Total of OTUs were assigned into 5 fungal phyla, 17 classes, 60 orders, 133 families, 195 genera, and 280 species. Ascomycota was the dominant phylum with 75.11% relative abundance in all samples, followed by Basidiomycota with 5.28%, while Kickxellomycota, Mortierellomycota, Mucoromycota, and other fungi had very low relative abundance of 0.01, 0.02, 0.07, and 0.01% respectively. In the total, 19.50% of fungal OTUs remained unidentified. Leptosillia, Aureobasidium and Acanthostigma were the most abundant genera detected in Q. serrata with the average relative abundance of 2.8, 2.76, and 2.19%, respectively. On the other hand, Peltaster, Cladosporium and Monochaetia were the most common genera detected in Q. mongolica with the average relative abundance of 4.83, 3.03, and 2.87%, respectively.
Supplemental Material
Download MS Excel (86.5 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- Gouda S, Das G, Sen SK, et al. Endophytes: a treasure house of bioactive compounds of medicinal importance. Front Microbiol. 2016;7:1538.
- Sun X, Guo LD. Endophytic fungal diversity: review of traditional and molecular techniques. Mycology. 2012;3(1):65–76.
- Santoyo G, Moreno-Hagelsieb G, Orozco-Mosqueda MDC, et al. Plant growth-promoting bacterial endophytes. Microbiol Res. 2016;183:92–99.
- Miliute I, Buzaite O, Baniulis D, et al. Bacterial endophytes in agricultural crops and their role in stress tolerance: a review. Zemdirb-Agric. 2015;102(4):465–478.
- Thomas T, Gilbert J, Meyer F. Metagenomics – a guide from sampling to data analysis. Microb Inform Exp. 2012;2(1):3.
- Streit WR, Schmitz RA. Metagenomics - the key to the uncultured microbes. Curr Opin Microbiol. 2004;7(5):492–498.
- Riesenfeld CS, Schloss PD, Handelsman J. Metagenomics: genomic analysis of microbial communities. Annu Rev Genet. 2004;38(1):525–552.
- Jumpponen A, Jones KL. Massively parallel 454 sequencing indicates hyperdiverse fungal communities in temperate Quercus macrocarpa phyllosphere. New Phytol. 2009;184(2):438–448.
- Jumpponen A, Jones KL. Seasonally dynamic fungal communities in the Quercus macrocarpa phyllosphere differ between urban and nonurban environments. New Phytol. 2010;186(2):496–513.
- Wemheuer F, Wemheuer B, Daniel R, et al. Deciphering bacterial and fungal endophyte communities in leaves of two maple trees with green islands. Sci Rep. 2019;9(1):14183.
- Gweon HS, Oliver A, Taylor J, et al. PIPITS: an automated pipeline for analyses of fungal internal transcribed spacer sequences from the Illumina sequencing platform. Methods Ecol Evol. 2015;6(8):973–980.
- Kemler M, Garnas J, Wingfield MJ, et al. Ion Torrent PGM as tool for fungal community analysis: a case study of endophytes in Eucalyptus grandis reveals high taxonomic diversity. PLOS One. 2013;8(12):e81718.
- Whiteley AS, Jenkins S, Waite I, et al. Microbial 16S rRNA Ion Tag and community metagenome sequencing using the Ion Torrent (PGM) platform. J Microbiol Methods. 2012;91(1):80–88.
- Paul NC, Yu SH. Endophytic fungi from medicinal plants in Korea. Saarbrücken, Germany: Lap Lambert Academic Publishing; 2011.
- Kim CK, Eo JK, Eom AH. Diversity and seasonal variation of endophytic fungi isolated from three conifers in Mt. Taehwa, Korea. Mycobiology. 2013;41(2):82–85.
- Min YJ, Park MS, Fong JJ, et al. Diversity and saline resistance of endophytic fungi associated with Pinus thunbergii in coastal shelterbelts of Korea. J Microbiol Biotechnol. 2014;24(3):324–333.
- You YH, Park JM, Seo YG, et al. Distribution, characterization, and diversity of the endophytic fungal communities on Korean seacoasts showing contrasting geographic conditions. Mycobiology. 2017;45(3):150–159.
- Eo JK, Park H, Eom AH. Diversity of endophytic fungi isolated from Pinus densiflora and Juniperus rigida distributed in Mt. Baekryeonsan and Mt. Johangsan, Korea. Korean J Mycol. 2018;46(4):437–446.
- Eo JK, Eom AH. Diversity of foliar endophytic fungi inhabiting coniferous trees in Korea. Korean J Mycol. 2018;46(3):205–211.
- Jung JY, Lee SH, Kim JM, et al. Metagenomic analysis of Kimchi, a traditional Korean fermented food. Appl Environ Microbiol. 2011;77(7):2264–2274.
- Oh SY, Fong JJ, Park MS, et al. Identifying airborne fungi in Seoul, Korea using metagenomics. J Microbiol. 2014;52(6):465–472.
- Cha S, Srinivasan S, Jang JH, et al. Metagenomic analysis of airborne bacterial community and diversity in Seoul, Korea, during December 2014, Asian dust event. PLOS One. 2017;12(1):e0170693.
- Kim H, Kim H, Hwang HS, et al. Metagenomic analysis of the marine coastal invertebrates of South Korea as assessed by Ilumina MiSeq. Anim Cells Syst. 2017;21(1):37–44.
- You Y, Kwon EJ, Choi S, et al. Vaginal microbiome profiles of pregnant women in Korea using a 16S metagenomics approach. Am J Reprod Immunol. 2019;82(1):e13124.
- Zhao CC, Eun JB. Shotgun metagenomics approach reveals the bacterial community and metabolic pathways in commercial Hongeo product, a traditional Korean fermented skate product. Food Res Int. 2020;131:109030.
- Kim CS, Jo JW, Lee H, et al. Comparison of soil higher fungal communities between dead and living Abies koreana in Mt. Halla, the Republic of Korea. Mycobiology. 2020;48(5):364–372.
- Kubono T, Ito S. Raffaelea quercivora sp. nov. associated with mass mortality of Japanese oak, and the ambrosia beetle (Platypus quercivorus). Mycoscience. 2002;43(3):255–260.
- Kim KH, Choi YJ, Seo ST, et al. Raffaelea quercus-mongolicae sp. nov. associated with Platypus koryoensis on oak in Korea. Mycotaxon. 2009;110(1):189–197.
- Toju H, Tanabe AS, Yamamoto S, et al. High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PLoS One. 2012;7(7):e40863.
- Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–2963.
- Li W, Fu L, Niu B, et al. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief Bioinform. 2012;13(6):656–668.
- Li W, Chang Y. CD-HIT-OTU-MiSeq, an improved approach for clustering and analyzing paired end MiSeq 16S rRNA sequences. BioRxiv. 2017. [Preprint]. Available from: https://www.biorxiv.org/content/early/2017/06/22/153783
- Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336.
- Abarenkov K, Nilsson HR, Larsson KH, et al. The UNITE database for molecular identification of fungi-recent updates and future perspectives. New Phytol. 2010;186(2):281–285.
- R Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2020.
- Zhao Y, Xiong Z, Wu G, et al. Fungal endophytic communities of two wild rosa varieties with different powdery mildew susceptibilities. Front Microbiol. 2018;9:2462.
- Moler ERV, Aho K. Whitebark pine foliar fungal endophyte communities in the Southern Cascade Range, USA: host mycobiomes and white pine blister rust. Fungal Ecol. 2018;33:104–114.
- Unterseher M, Peršoh D, Schnittler M. Leaf-inhabiting endophytic fungi of European beech (Fagus sylvatica L.) co-occur in leaf litter but are rare on decaying wood of the same host. Fungal Divers. 2013;60(1):43–54.
- Abdelfattah A, Wisniewski M, Nicosia MGLD, et al. Metagenomic analysis of fungal diversity on strawberry plants and the effect of management practices on the fungal community structure of aerial organs. PLOS One. 2016;11(8):e0160470.
- Kurandawad JM, Lakshman HC. Diversity of the endophytic fungi isolated from Acalypha indica Linn – a promising medicinal plant. Int J Sci Res Publ. 2014;4(4):1–7.
- Mishra A, Gond SK, Kumar A, et al. Season and tissue type affect fungal endophyte communities of the Indian medicinal plant Tinospora cordifolia more strongly than geographic location. Microb Ecol. 2012;64(2):388–398.
- Park YH, Lee SG, Ahn DJ, et al. Diversity of fungal endophytes in various tissues of Panax ginseng Meyer cultivated in Korea. J Ginseng Res. 2012;36(2):211–217.
- Gazis R, Chaverri P. Diversity of fungal endophytes in leaves and stems of wild rubber trees (Hevea brasiliensis) in Peru. Fungal Ecol. 2010;3(3):240–254.
- Peršoh D, Melcher M, Flessa F, et al. First fungal community analyses of endophytic ascomycetes associated with Viscum album ssp. austriacum and its host Pinus sylvestris. Fungal Biol. 2010;114(7):585–596.
- Khan AR, Waqas M, Ullah I, et al. Culturable endophytic fungal diversity in the cadmium hyperaccumulator Solanum nigrum L. and their role in enhancing phytoremediation. Environ Exp Bot. 2017;135:126–135.
- Cleary M, Nguyen D, Marčiulynienė D, et al. Friend or foe? Biological and ecological traits of the European ash dieback pathogen Hymenoscyphus fraxineus in its native environment. Sci Rep. 2016;6(1):21895.
- Ragazzi A, Moricca S, Capretti P, et al. Endophytic fungi in Quercus cerris: isolation frequency in relation to phenological phase, tree health and the organ affected. Phytopathol Mediterr. 2001;40:165–171.
- He X, Han G, Lin Y, et al. Diversity and decomposition potential of endophytes in leaves of a Cinnamomum camphora plantation in China. Ecol Res. 2012;27(2):273–284.
- Bejarano NV, Carrillo L. Fungal endophytes in sweet orange Citrus sinensis (L.) Osbeck in Jujuy-Argentina. Asian J Agric Food Sci. 2016;4(1):54–59.
- Naik BS, Shashikala J, Krishnamurthy YL. Study on the diversity of endophytic communities from rice (Oryza sativa L.) and their antagonistic activities in vitro. Microbiol Res. 2009;164(3):290–296.
- Larran S, Perelló A, Simón MR, et al. The endophytic fungi from wheat (Triticum aestivum L.). World J Microbiol Biotechnol. 2007;23(4):565–572.
- Reiher DBA. Leaf-inhabiting endophytic fungi in the canopy of the Leipzig Floodplain Forest [Ph.D Thesis]. Germany: University of Leipzig; 2011.
- Davydenko K, Vasaitis R, Stenlid J, et al. Fungi in foliage and shoots of Fraxinus excelsior in Eastern Ukraine: a first report on Hymenoscyphus pseudoalbidus. For Path. 2013;43(6):462–467.
- Albrectsen BR, Björkén L, Varad A, et al. Endophytic fungi in European aspen (Populus tremula) leaves-diversity, detection, and a suggested correlation with herbivory resistance. Fungal Divers. 2010;41(1):17–28.
- Fisher PJ, Petrini O, Scott HML. The distribution of some fungal and bacterial endophytes in maize (Zea mays L.). New Phytol. 1992;122(2):299–305.
- Parsa S, García-Lemos AM, Castillo K, et al. Fungal endophytes in germinated seeds of the common bean, Phaseolus vulgaris. Fungal Biol. 2016;120(5):783–790.
- Powthong P, Jantrapanukorn B, Thongmee A, et al. Screening of antimicrobial activities of the endophytic fungi isolated from Sesbania grandiflora (L.) Pers. J Agric Sci Technol. 2013;15:1513–1522.
- Verma VC, Gond SK, Kumar A, et al. The endophytic mycoflora of bark, leaf, and stem tissues of Azadirachta indica A. Juss (neem) from Varanasi (India). Microb Ecol. 2007;54(1):119–125.
- Rateb ME, Ebel R. Secondary metabolites of fungi from marine habitats. Nat Prod Rep. 2011;28(2):290–344.
- Jiang Q, Wei N, Huo Y, et al. Secondary metabolites of the endophytic fungus Cladosporium sp. CYC38. Chem Nat Compd. 2020;56(6):1166–1169.
- Amirita A, Sindhu P, Swetha J, et al. Enumeration of endophytic fungi from medicinal plants and screeening of extracellular enzymes. World J Sci Tech. 2012;2(2):13–19.
- Patil MG, Pagare J, Patil SN, et al. Extracellular enzymatic activities of endophytic fungi isolated from various medicinal plants. Int J Curr Microbiol Appl Sci. 2015;4(3):1035–1042.
- Zhang D, Spadaro D, Garibaldi A, et al. Efficacy of the antagonist Aureobasidium pullulans PL5 against postharvest pathogens of peach, apple and plum and its modes of action. Biol Control. 2010;54(3):172–180.
- Mari M, Martini C, Guidarelli M, et al. Postharvest biocontrol of Monilinia laxa, Monilinia fructicola and Monilinia fructigena on stone fruit by two Aureobasidium pullulans strains. Biol Control. 2012;60(2):132–140.
- Francesco AD, Foggia MD, Baraldi E. Aureobasidium pullulans volatile organic compounds as alternative postharvest method to control brown rot of stone fruits. Food Microbiol. 2020;87:103395.
- Schena L, Nigro F, Pentimone I, et al. Control of postharvest rots of sweet cherries and table grapes with endophytic isolates of Aureobasidium pullulans. Postharvest Biol Technol. 2003;30(3):209–220.
- Francesco AD, Ugolini L, Lazzeri L, et al. Production of volatile organic compounds by Aureobasidium pullulans as a potential mechanism of action against postharvest fruit pathogens. Biol Control. 2015;81:8–14.
- Francesco AD, Zajc J, Gunde-Cimerman N, et al. Bioactivity of volatile organic compounds by Aureobasidium species against gray mold of tomato and table grape. World J Microbiol Biotechnol. 2020;36(11):171.
- Agirman B, Erten H. Biocontrol ability and action mechanisms of Aureobasidium pullulans GE17 and Meyerozyma guilliermondii KL3 against Penicillium digitatum DSM2750 and Penicillium expansum DSM62841 causing postharvest diseases. Yeast. 2020;37(9-10):437–448.
- Francesco AD, Milella F, Mari M, et al. A preliminary investigation into Aureobasidium pullulans as a potential biocontrol agent against Phytophthora infestans of tomato. Biol Control. 2017;114:144–149.
- Miles LA, Lopera CA, González S, et al. Exploring the biocontrol potential of fungal endophytes from an Andean Colombian Paramo ecosystem. BioControl. 2012;57(5):697–710.
- Francesco AD, Foggia MD, Corbetta M, et al. Biocontrol activity and plant growth promotion exerted by Aureobasidium pullulans strains. J Plant Growth Regul. 2021;40:1233–1244.
- Wachowska U, Głowacka K. Antagonistic interactions between Aureobasidium pullulans and Fusarium culmorum, a fungal pathogen of winter wheat. BioControl. 2014;59(5):635–645.
- Rusin C, Francesco AD, Foggia MD, et al. An emerging problem affecting apple production: Neofusicoccum parvum. Aureobasidium pullulans L1 and L8 strains as an alternative control strategy. Biol Control. 2019;134:157–162.
- Andreea C, Giménez-Mariño C, Cabrera Y, et al. Endophytic fungi from grapevine cultivars in Canary Islands and their activity against phytopatogenic fungi. Int J Agric Crop Sci. 2014;7(15):1497–1503.
- Khan AL, Al-Harrasi A, Al-Rawahi A, et al. Endophytic fungi from frankincense tree improves host growth and produces extracellular enzymes and indole acetic acid. PLoS One. 2016;11(6):e0158207.
- Cho SE, Lee DH, Wingfield MJ, et at. Ceratocystis quercicola sp. nov. from Quercus variabilis in Korea. Mycobiology. 2020;48(4):245–251.