
Abstract
The ascomycete fungus Cordyceps militaris infects lepidopteran larvae and pupae and forms characteristic fruiting bodies. Owing to its immune-enhancing effects, the fungus has been used as a medicine. For industrial application, this fungus can be grown on geminated soybeans as an alternative protein source. In our study, we performed a comprehensive transcriptomic analysis to identify core gene sets during C. militaris cultivation on germinated soybeans. RNA-Seq technology was applied to the fungal cultures at seven-time points (2, 4, and 7-day and 2, 3, 5, 7-week old cultures) to investigate the global transcriptomic change. We conducted a time-series analysis using a two-step regression strategy and chose 1460 significant genes and assigned them into five clusters. Characterization of each cluster based on Gene Ontology and Kyoto Encyclopedia of Genes and Genomes databases revealed that transcription profiles changed after two weeks of incubation. Gene mapping of cordycepin biosynthesis and isoflavone modification pathways also confirmed that gene expression in the early stage of GSC cultivation is important for these metabolic pathways. Our transcriptomic analysis and selected genes provided a comprehensive molecular basis for the cultivation of C. militaris on germinated soybeans.
1. Introduction
The ascomycete fungus Cordyceps militaris can infect lepidopteran pupae, which includes over 500 species capable of infecting many other insects, spiders, and mites [Citation1]. When this fungus colonizes dead or living Lepidoptera caterpillars, the mycelium infiltrates into the caterpillar body and finally replaces most host tissues. Later, fruiting bodies are formed on the surface of the insects’ body. They are called “Dong Choong Ha Cho” in Korea, meaning that worms in winter turn into plants in summer. The fruiting bodies have been used as medicine in Asia for a long time [Citation2]. Anti-tumor, anti-inflammatory, and anti-oxidative activities have been reported in the fruiting bodies of C. militaris [Citation3–8]. One of the main bioactive ingredients of C. militaris, 3′-deoxyadenosine (also called cordycepin), displays anti-tumor activity via an apoptosis mechanism [Citation9–11].
The genome of C. militaris has been estimated as 32.27 Mb and has 9810 genes [Citation12]. After the publication of its whole genome sequence, possible metabolic pathways for cordycepin biosynthesis have bezen suggested based on bioinformatic data [Citation12,Citation13]. Recently, a reverse genetics approach with liquid chromatography was applied to identify a cordycepin/pentostatin biosynthesis cluster, which consists of four genes (named CNS1-4) [Citation14]. In that study, CNS1 and CNS2 genes were indispensable for cordycepin biosynthesis, and CNS3 was essential for pentostatin biosynthesis. The function of CNS4 was predicted to be the transport of pentostatin to balance the intracellular concentration of cordycepin [Citation14]. In addition, many transcriptome studies have been performed using C. militaris, focusing on fruiting body formation [Citation12,Citation15] and optimization of cordycepin production [Citation16–18]. Recently, many comparative transcriptomic studies have suggested metabolic pathways and key enzymes involved in cordycepin production. These studies demonstrated that it is practicable to improve the productivity of cordycepin through analysis and regulation of its biosynthetic pathways [Citation15,Citation19–21]. Despite many studies having been conducted on the biosynthetic process of cordycepin in C. militaris, there is no report on the biosynthetic process of other useful bioactive ingredients, such as isoflavones at the level of gene expression in C. militaris.
Although fruiting bodies of C. militaris have medicinal effects on human health, it is difficult to produce them on a large scale because of the difficulty in culturing this fungus on insect pupae. To overcome this problem, a method was developed in which germinated soybeans were used as a protein source instead of pupae for artificial mass cultivation [Citation22]. In this method, the mycelia of C. militaris were placed on sterile germinated soybeans and incubated at 25 °C for 8 weeks. This solid culture is hereinafter referred to as GSC. Metabolomics and multivariate statistical analyses were conducted to compare compositional changes between germinated soybean (GS) and GSC [Citation23]. One-week-old cultures exhibited strong GSC biological activity. Interestingly, new forms of isoflavones, a modified version of genistein and daidzein, were detected in GSCs. The isoflavones significantly inhibited degranulation in antigen-stimulated RBL-2H3 cells, suggesting that GSC is an anti-allergic agent for treating IgE-mediated allergic disease [Citation24]. Ethanol extracts of GSC showed anti-inflammatory effects on acute colitis symptoms [Citation25]. These results reveal the efficiency of GSC, which can be used as a promising drug or health supplement.
In this study, we conducted time-series gene expression profiling during GSC cultivation to identify marker genes at different culture stages and to map those genes to the biosynthesis pathways of cordycepin or modified isoflavones.
2. Materials and methods
2.1. Fungal isolates
The C. militaris isolate KUCARI_0903 was obtained from the Cell Activation Research Institute (Seoul, Korea). The culture was maintained on a potato dextrose agar plate at 25 °C in the dark.
2.2. Fungal culture on soybeans and its sampling
Three-day-old C. militaris culture was filtered through a three-layered cheese cloth to remove potato dextrose broth. The fungal mycelia were ground using a sterile spoon and cloth, and 25 ml of sterile distilled water was added. Germinated soybeans (Glycine max, “Jui Nun Yi Cong”) were autoclaved and placed in a 30-ml plate (Fisher Scientific International, Inc., Hampton, USA) (). Four milliliters of mycelia ground with water was used as the inoculum for each plate. The mycelia on the germinated soybeans were cultured at 25 °C for 8 weeks () [Citation22,Citation23]. For RNA-Seq, fungal mycelia were sampled at 2, 4, and 7 days post-inoculation (dpi) and 2, 3, 5, and 7 weeks post-inoculation (wpi). Each sample harvested from three plates was pooled for cost-saving before RNA extraction [Citation26].
Figure 1. Soybeans with Cordyceps militaris. (A) Soybeans (Glycine max, “Jui Nun Yi Cong” in Korean); (B) Germinated soybeans; (C) C. militaris mycelia grown on the germinated soybeans for eight weeks; (D) C. militaris growing on the germinated soybeans (GSC).

2.3. Extraction of RNA and RNA-sequencing
Total RNA was extracted using the TRIzol® reagent (Life Technologies, MD, USA). RNA quality was confirmed using an Agilent 2100 bioanalyzer using the RNA 6000 Nano Chip (Agilent Technologies, Amstelveen, The Netherlands), and RNA quantification was performed using an NA-2000 Spectrophotometer (Thermo Fisher Scientific Inc., DE, USA). The TruSeq™ RNA sample preparation kit (Illumina, CA, USA) was used for mRNA-seq library construction, following the manufacturer’s instructions. High-throughput sequencing was performed as paired-end 100 sequencing using the Illumina HiSeq 2500 platform (Illumina, Inc., USA). The high-quality sequences were deposited in the NCBI Sequence Read Archive database with the accession no SAMN22563802-8 and PRJNA774439.
2.4. Read filtering, mapping, and expression quantification
Paired-end reads were filtered using a Flexbar (ver. 2.5). The program discarded uncalled bases, adaptor sequences, and short reads. HISAT (ver. 2.0.4) [Citation27] was used to map the filtered reads against the reference genome of C. militaris [Citation12]. The genome was 32.2 Mb in size and contained 9651 coding and 159 non-coding genes (C. militaris strain CM01 ver. 1.38; http://fungi.ensembl.org).
Differentially expressed genes were determined based on counts from unique and multiple alignments using coverage in Bedtools [Citation28]. The read count data were processed based on the quantile normalization method using EdgeR using Bioconductor [Citation29]. The alignment files were also used for assembling transcripts, estimating their abundances, and detecting differential expression of genes or isoforms using cufflinks. We used the fragments per kilobase of exon per million fragments (FPKM) to determine the expression level of the gene regions.
The count data were imported using “DESeqDataSetFromHTSeq” implemented in the tximport package (ver. 1.11.7). Raw counts were normalized using the DESeq2 package (ver. 1.22.2). Time-series analysis was performed using the maSigPro package (ver. 1.54.0). The p-values were adjusted for multiple comparisons using false discovery rate (FDR). We used “degree = 2” for three time points or “degree = 3” for four time points in the function “make.design.matrix.” The cutoff value for the R-squared value of the regression model was set as 0.8.
2.5. Gene ontology
A weight algorithm with Fisher’s exact tests implemented in the R package “topGO” version 2.12.0 was employed to calculate the GO term significance for enrichment analysis of gene sets and visualization of a GO hierarchical structure. GO terms for genes of C. militaris were retrieved from the Gene Ontology Annotation database (http://www.ebi.ac.uk/GOA/).
2.6. Real-time quantitative reverse transcription PCR (qRT-PCR)
RNA-Seq data were confirmed by qRT-PCR. Complementary DNA was prepared from total RNA using ImProm-II™ Reverse Transcription System (Promega, Madison, WI, USA) according to the manufacturer’s recommendation. PCR reactions were carried out in the mixture of cDNA template (25 ng/2 µl), reverse and forward primers (100 nM each in total 3 µl) and Power SYBR® Green PCR Master Mix (5 µl, Applied Biosystems, Foster City, CA, USA). The AB7500 Real-Time PCR system was used for amplification with the following condition: 40 cycles of 15 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C after initial denaturation (Applied Biosystems). The beta-tubulin gene was used as an endogenous control. Primers are listed in Table S1.
3. Results and discussion
3.1. Overall similarity between samples
Cordyceps militaris was inoculated on GSCs and cultivated for eight weeks (). We chose seven time points to investigate the global transcriptome of GSCs: 2, 4, 7 days, 2, 3, 5, and 7 weeks post-inoculation. Total RNA from seven GSC samples was isolated and sequenced using the Illumina HiSeq platform. After filtration of the reads containing adaptor or low-quality sequences, a total of 205 million paired-end reads were obtained. Approximately 75% of the filtered reads were mapped to the genome of the C. militaris strain CM01 (ver. 1.38; http://fungi.ensembl.org). As shown in , 60.8% of the mapped reads were aligned uniquely to the genomic location on average.
Table 1. Summary of RNA-seq data.
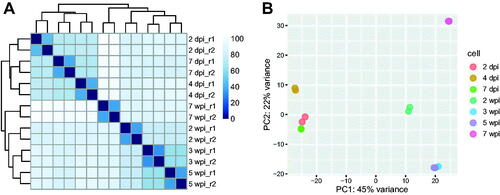
To assess the overall similarity between samples, a regularized logarithm transformation of the count data was applied [Citation30]. The 6727 genes with at least ten counts across all samples were used for further analyses. A heatmap was generated based on the Euclidean distance between samples (). On the heatmap, the average distance between samples was 86.2 while the distance between replicates was 38.2. The day samples (2, 4, and 7 dpi) were clustered together with an average distance of 72.3, and apart from the week samples (2, 3, 5, and 7 wpi) with an average distance of 92.2. Additionally, a principal component analysis (PCA) showed similarity as a heatmap (). The day samples were far from the week samples. The “2 and 7 dpi” and “3 and 5 wpi” samples were located very close to each other. However, the “7 wpi” sample was far from the other samples based on the PC2 axis ().
Figure 2. Overall similarity of each sample using a regularized-logarithm transformation. (A) Sample distance matrix using heatmap with Euclidean distance of 6727-gene transcriptome of each sample; (B) Principal component analysis of 6727-gene transcriptome of each sample.
3.2. Clustering of differentially expressed genes
Time-series differential expression analysis was performed to examine the global transcriptomic patterns of the seven GSC samples. The two-step regression strategy in the maSigPro R package was used to find a gene set with differential expression [Citation31]. A total of 1460 significant genes were selected using the following options (degree = 4, Q = 0.05, theta = 5, and rsq = 0.7) and classified into five co-expressed gene clusters (k = 5). Of the significant genes, 181, 311, 196, 332, and 440 genes were assigned to clusters I–V, respectively (). The expression pattern of 10 genes in the clusters was confirmed using qRT-PCR (Figure S1). Two genes in each cluster Clusters I and II contained genes induced within 1 week, while genes in clusters III, IV, and V showed higher expression at later incubation (2–7 wpi). Interestingly, cluster V was the largest group in size and had highly expressed genes only at 7 wpi (). This explains why the “7 wpi” sample is different from the others in the PCA result ().
Figure 3. Gene cluster analysis and enrichment of GO and KEGG pathways for the 1460 significant genes according to a time-series differential expression. Heatmaps, gene ontology, and KEGG pathways of genes belonging to each cluster. In total, 181, 311, 196, 332, and 440 genes were assigned as cluster I–V, respectively.
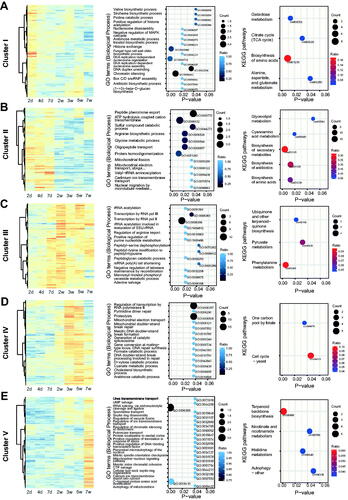
GO and KEGG enrichment analyses were performed for each gene cluster. In cluster I, 17 GOs were identified as significant from the 181 identified DEGs (, Table S2). Among them, eight GOs were related to chromatin remodeling or reorganization of nucleosomes (GO:0000492, GO:0006335, GO:0006337, GO:0006342, GO:0032508, GO:0034724, GO:0035066, and GO:0043486). Given that the gene expression in cluster I peaked at 2 dpi, the fungal cells might make the transition from a nutrient-rich environment (i.e., PDA) to a new and poor environment (i.e., soybean surface) by remodeling chromatin structure. In particular, the histone chaperone (CCM_05963, ASF1), a component of the FACT complex (CCM_05775, POB3), and RuvB-like DNA helicase genes participated in chromatin remodeling. Additionally, biosynthesis of amino acids (GO: 0006021, GO:0006562, GO: 0009099, and GO: 0046373) and cell wall components (GO:0006075 and GO:0034221) were activated, indicating that this fungus was producing basic cell metabolites to grow on the germinated soybeans. All the KEGG pathways enriched in cluster I were involved in carbohydrate and amino acid metabolism (galactose metabolism, citrate cycle, biosynthesis of amino acids, alanine aspartate, and glutamate metabolism; Table S3).
Cluster II consisted of 311 DEGs that showed strong expression within one week. Five out of 12 GOs in this cluster were involved in both energy and amino acid metabolism (GO:0099132, GO:0006122, GO:0006526, GO:0006544, and GO:0044273, Table S2). Two out of five KEGG pathways were involved in amino acid metabolism (cmt01230 and cmt00460). Thus, genes involved in the primary metabolism of energy, amino acids, and carbohydrates were included mainly in clusters II and I. This indicates that fungal growth on the germinated soybeans was active within one week of incubation on germinated soybean. Interestingly, the antibiotic (or secondary metabolite) biosynthetic pathway (cmt01130 and cmt01110) was significantly highlighted in cluster II (). The ABC multidrug transporter CCM_07735 is one of the genes involved in this pathway. This gene was highly expressed in the aerial mycelia of C. militaris compared to the submerged incubation condition [Citation32].
Cluster III contained 196 DEGs that were continuously expressed throughout the incubation after one week (). Thus, these genes may be good genetic markers for GSC cultivation. For example, the CCM_04506 gene displayed 100-fold higher expression even in all selection points, compared to the “0 day” control. In cluster III, six of the 13 GO terms were related to transcription and translation (GO:0006366, GO:0006383, GO:0006168, GO:0051391, GO:1904812, and GO:0000289) (). Three KEGG pathways, phenylalanine metabolism, ubiquinone, and another terpenoid-quinone biosynthesis, and pyruvate metabolism (cmt00369, cmt00130, and cmt00620, respectively), were highlighted. Two genes, CCM_00270 and CCM_06000, which were involved in 4-hydroxyphenylpyruvate dioxygenase activities, were found in two KEGG pathways (cmt00369 and cmt00130) in common. The GO and KEGG analyses indicated that the gene expressions for catabolic and anabolic metabolism were active in this cluster.
The genes in cluster IV were up-regulated after two weeks. The biggest GO term in this cluster was related to transcription (GO:0006357, regulation of transcription by RNA polymerase II) (). This is consistent with the enriched GO in cluster III. Among the 14 GO terms, five were involved in DNA repair (GO:0000734, GO:0006290, GO:0010780, GO:0010791, and GO:0097552). In particular, three of these GO terms were enriched due to DNA repair protein rad32 (CCM_2130), which belongs to the MRE11/RAD32 family. Likewise, “cell cycle” (cmt04111) pathway was enriched from the KEGG pathway analysis although a different set of genes contributed to the enrichment of this pathway (CCM_00489, CCM_01153, CCM_05144, CCM_07049, and CCM_07539 in Table S3).
Cluster V was the largest among the five clusters in terms of the number of DEGs (440 DEGs), where 21 GO terms were highlighted (). Contrary to the cluster size in this cluster, GO-annotated genes were rare in this cluster. The most enriched GO term was “tRNA splicing, via endonucleolytic cleavage and ligation” (GO: 0006388). This term consisted of four genes, three of which (CCM_00392, CCM_00753, and CCM_09522) belonged to cluster V. The second biggest GO term is “amino acid modification” (GO:0018410). This term contains two DEGs, protein-S-isoprenylcysteine O-methyltransferase (CCM_05030) and autophagy-related protein 3 (CCM_06512). CCM_6512 contributed to two autophagy-related GO terms (GO:0000422 and GO:0034727). Consistently, one of the KEGG pathways was the autophagy pathway (cmt04136), where three genes in cluster V were involved (CCM_00278, CCM_00551, and CCM_06512). The WD-repeat protein (CCM_00551) also contributed to the mitochondria-nucleus signaling pathway (GO:0031930, Table S2). The last two clusters (IV and V) have many genes involved in DNA repair, proteolysis, and autophagy, suggesting that the culture turns into the death phase where dead cells are more than living cells.
3.3. Gene expression of cordycepin biosynthesis pathway
Cordycepin, a structural derivative of adenosine, is produced in C. militaris. During GSC cultivation, we found that 180.58 ± 1.54 mg/g cordycepin was produced. Recently, one of the biosynthetic pathways was confirmed using mutagenesis and metabolomics approaches [Citation14]. The characterized pathways and genes are shown in . In summary, the precursor adenosine is converted into adenosine-3′-monophosphate (3′-AMP) by the activity of the phosphohydrolase (Cns3, CCM_04438) NK domain. Phosphohydrolase (Cns2, CCM_04437) catalyzes 3′-AMP into 2′-carbonyl-3′-deoxyadenosine (2′-C-3′-dA), which is further converted to cordycepin by oxidoreductase (Cns1, CCM_04436) through a reduction reaction. When cordycepin accumulated within the cell at the toxic level, pentostatins are pumped out through the ABC transporter (Cns4, CCM_04439) and pentostatin-suppressed deaminases convert cordycepin into the less toxic molecule, 3′-deoxyinosine. We mapped our transcriptome data to this pathway (). Among the four genes, CNS1, CNS2, and CNS3 were strongly expressed at most cultivation times (average regularized-logarithm values were 3.9, 4.0, and 4.9, respectively). While the expression of CNS3 was highest at 2 dpi, both CNS1 and CNS2 were the highest at 7 dpi. This time lag makes sense because adenosine is a precursor of cordycepin. The expression of CNS2 and CNS3 decreased at 7 wpi. Interestingly, two candidate genes for cordycepin deamination, CCM_02911 and CCM_09449, were upregulated at 7 wpi (both belong to cluster V). The patterns of their expressions were opposite to those of CNS2 and CNS3, providing two insights. First, reduction of gene expression reduces the production of both pentostatin and cordycepin directly, lowering intracellular levels. Second, a common transcriptional regulator might contribute to a sudden decrease in the transcripts of CNS2/CNS3 and, at the same time, a sudden increase in the expression of CCM_02911/CCM_09449. In agreement with Xia et al. [Citation14], the CNS4 gene might also function as a suppressor of inactivation through the transport of pentostatin. It showed weak expression during most cultivation times, compared to the other genes (the average regularized-logarithm value was 0.9). However, its expression increased at 3 and 7 wpi, which might increase the out-pumping of pentostatins and activate the deaminases.
Figure 4. Cordycepin biosynthetic pathways. The putative pathway is based on the study by Xia et al. [Citation14]. The heatmap was generated from the regularized-logarithm data after subtraction of the mean expression for each gene. Cns1 (CCM_04436): oxidoreductase, Cns2 (CCM_04437): phosphohydrolases, Cns3 (CCM_04438): ATP phosphoribosyltransferases (HisG and NK are domain names), Cns4 (CCM_04439): ATP-binding cassette transporter, CCM_02911: adenosine deaminase, CCM_09449: adenosine deaminase.
![Figure 4. Cordycepin biosynthetic pathways. The putative pathway is based on the study by Xia et al. [Citation14]. The heatmap was generated from the regularized-logarithm data after subtraction of the mean expression for each gene. Cns1 (CCM_04436): oxidoreductase, Cns2 (CCM_04437): phosphohydrolases, Cns3 (CCM_04438): ATP phosphoribosyltransferases (HisG and NK are domain names), Cns4 (CCM_04439): ATP-binding cassette transporter, CCM_02911: adenosine deaminase, CCM_09449: adenosine deaminase.](/cms/asset/93a63be3-1091-4277-af94-e11884ea3b65/tmyb_a_2035906_f0004_c.jpg)
In addition, Vongsangnak et al. predicted diverse metabolic pathways in C. militaris using a comparative genomic approach [Citation13]. Among the 1267 metabolic reactions, 29 metabolic reactions consisting of 37 genes were related to cordycepin biosynthesis [Citation13]. Eight of the thirty seven genes matched with the 1460 significant genes (CCM_00088, CCM_02051, CCM_02911, CCM_05543, CCM_06062, CCM_07145, CCM_09067, and CCM_09449 in Table S4). Seven of the genes were included in clusters III to V, suggesting that these genes could participate in cordycepin biosynthesis late during GSC cultivation. PCA with the 37 genes exhibited separation of “7 wpi” from the other samples (). Interestingly, the expression of two deaminases (CCM_02911 and CCM_09449) uniquely contributed to the separation of “7 wpi.”
Figure 5. Principal component analysis for the 37 genes involved in cordycepin biosynthesis. The plot was generated from the regularized logarithm values for the count data. The 37 genes were chosen based on the metabolic pathways constructed by Vongsangnak et al. [Citation13]. The eight genes matched to the 1460 significant genes are indicated with rounded rectangles.
![Figure 5. Principal component analysis for the 37 genes involved in cordycepin biosynthesis. The plot was generated from the regularized logarithm values for the count data. The 37 genes were chosen based on the metabolic pathways constructed by Vongsangnak et al. [Citation13]. The eight genes matched to the 1460 significant genes are indicated with rounded rectangles.](/cms/asset/cda9dc8a-87ac-41df-b89a-aa04b2ca4dc4/tmyb_a_2035906_f0005_c.jpg)
3.4. Comparison with metabolomics of GSC cultivation
Previously, metabolomic changes in GSC cultivation were examined using both liquid chromatography and gas chromatography-mass spectrometry [Citation23]. The total flavonoid and phenolic contents (TFC and TPC) in GSC extracts increased after co-cultivation with C. militaris, suggesting that GSC showed higher antioxidant activities than germinated soybean only. Isoflavones, such as genistein, daidzein, and glycitein, are major secondary metabolites of soybean and are known to have strong antioxidant effects [Citation33,Citation34]. In nature, isoflavones are coupled with β-D-glucosides, which are called genistin, daidzin, and glycitin, respectively. They are synthesized through a phenylpropanoid pathway where cinnamic acid, p-coumaric acid, and liquiritigenin are produced in the order of phenyl alanine [Citation35]. Among the genes related to the phenylpropanoid pathway, only two genes, cytochrome P450 oxidoreductase (similar to trans-cinnamate 4-oxygenase, CCM_01530) and glycerol dehydrogenase (similar to calcone reductase, CCM_01682), were found in the C. militaris genome (), indicating that this fungus alone cannot complete isoflavone biosynthesis. In addition, several microbial or plant enzymes involved in the metabolic modification of isoflavones (hydrolysis, hydroxylation, reduction, and methylation) have been reviewed [Citation36]. In , we compared the key enzymes in the review of the C. militaris genome and transcriptome data. For example, β-glucosidase from Bifidobacterium pseudocatenulatum (Uniprot id: A0A072MX70) hydrolyzes glycosyl isoflavones into aglycones and isoflavones without attached glycosides [Citation37]. C. militaris has several orthologous genes (CCM_06730, CCM_05167, CCM_02750, and CCM_07910) in its genome, three of which were found in the clusters (). Methylation is also important in the modification of isoflavones, changing the solubility and biological activity [Citation38]. Flavonoid 4′-O-methyltransferase (Uniprot id: C6TAY1) had orthologous genes in C. militaris (CCM_01923, CCM_07823, CCM_05265, and CCM_04907). One of them (CCM_04907, O-methyltransferase) was selected in cluster II (). During GSC cultivation, new forms of isoflavones were reported (daidzein 7-O-β-D-glucoside 4″-O-methylate, glycitein 7-O-β-D-glucoside 4″-O-methylate, genistein 7-O-β-D-glucoside 4″-O-methylate, and genistein 4′-O-β-D-glucoside 4″-O-methylate) [Citation23]. For the reason that these new isoflavones have modifications at the isoflavone glycosides instead of those at the main isoflavones, it is still possible that other enzymes might be involved in the modification during GSC cultivation.
Table 2. The genes involved in phenylpropanoid pathway and isoflavone modification.
4. Conclusion
In this study, global transcriptional profiling was conducted during 8-week cultivation of C. militaris on germinated soybeans. Time-series transcriptome data revealed that this fungus showed active growth up to 7 dpi and autophagic status at 7 wpi. The genes related to cordycepin biosynthesis were highly expressed, supporting cordycepin production at the transcript level. Our data also support a putative controlling pathway. Isoflavone modification is unique to GSC cultivation Although genes involved in the modification have not yet been characterized, some candidate genes were predicted bioinformatically. Their expression was confirmed. Our transcriptomic analysis offers a comprehensive understanding of fungal responses to germinated soybeans and provides an idea to obtain bioactive ingredients effectively.
Supplemental Material
Download MS Excel (18.1 KB)Supplemental Material
Download MS Excel (45.3 KB)Supplemental Material
Download MS Excel (53.3 KB)Supplemental Material
Download MS Word (14.3 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Sung G-H, Hywel-Jones NL, Sung J-M, et al. Phylogenetic classification of Cordyceps and the Clavicipitaceous fungi. Stud Mycol. 2007;57:5–59.
- Paterson RRM. Cordyceps – a traditional Chinese medicine and another fungal therapeutic biofactory? Phytochemistry. 2008;69(7):1469–1495.
- Qin P, Li X, Yang H, et al. Therapeutic potential and biological applications of cordycepin and metabolic mechanisms in cordycepin-producing fungi. Molecules. 2019;24(12):2231.
- Jędrejko KJ, Lazur J, Muszyńska B. Cordyceps militaris: an overview of its chemical constituents in relation to biological activity. Foods. 2021;10(11):2634.
- Zhong J-J, Xiao J-H. Secondary metabolites from higher fungi: discovery, bioactivity, and bioproduction. In: Zhong J-J, Bai F-W, Zhang W, editors. Biotechnology in China I. Switzerland: Springer; 2009. p. 79–150.
- Noh E-M, Jung SH, Han J-H, et al. Cordycepin inhibits TPA-induced matrix metalloproteinase-9 expression by suppressing the MAPK/AP-1 pathway in MCF-7 human breast cancer cells. Int J Mol Med. 2010;25:255–260.
- Kim HG, Shrestha B, Lim SY, et al. Cordycepin inhibits lipopolysaccharide-induced inflammation by the suppression of NF-κB through Akt and p38 inhibition in RAW 264.7 macrophage cells. Eur J Pharmacol. 2006;545(2–3):192–199.
- Choi E, Oh J, Sung G-H. Antithrombotic and antiplatelet effects of Cordyceps militaris. Mycobiology. 2020;48(3):228–232.
- Shao LW, Huang LH, Yan S, et al. Cordycepin induces apoptosis in human liver cancer HepG2 cells through extrinsic and intrinsic signaling pathways. Oncol Lett. 2016;12(2):995–1000.
- Nasser MI, Masood M, Wei W, et al. Cordycepin induces apoptosis in SGC-7901 cells through mitochondrial extrinsic phosphorylation of PI3K/akt by generating ROS. Int J Oncol. 2017;50(3):911–919.
- Tian X, Li Y, Shen Y, et al. Apoptosis and inhibition of proliferation of cancer cells induced by cordycepin. Oncol Lett. 2015;10(2):595–599.
- Zheng P, Xia Y, Xiao G, et al. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2011;12(11):R116.
- Vongsangnak W, Raethong N, Mujchariyakul W, et al. Genome-scale metabolic network of Cordyceps militaris useful for comparative analysis of entomopathogenic fungi. Gene. 2017;626:132–139.
- Xia Y, Luo F, Shang Y, et al. Fungal cordycepin biosynthesis is coupled with the production of the safeguard molecule pentostatin. Cell Chem Biol. 2017;24(12):1479–1489.
- Yin Y, Yu G, Chen Y, et al. Genome-wide transcriptome and proteome analysis on different developmental stages of Cordyceps militaris. PLOS One. 2012;7(12):e51853.
- Yin J, Xin XD, Weng YJ, et al. Transcriptome-wide analysis reveals the progress of Cordyceps militaris subculture degeneration. PLOS One. 2017;12(10):e0186279.
- Raethong N, Laoteng K, Vongsangnak W. Uncovering global metabolic response to cordycepin production in Cordyceps militaris through transcriptome and genome-scale network-driven analysis. Sci Rep. 2018;8(1):1–13.
- Chen B-X, Wei T, Xue L-N, et al. Transcriptome analysis reveals the flexibility of cordycepin network in Cordyceps militaris activated by l-alanine addition. Front Microbiol . 2020;11:577.
- Xia F, Liu Y, Shen G-R, et al. Investigation and analysis of microbiological communities in natural Ophiocordyceps sinensis. Can J Microbiol. 2015;61(2):104–111.
- Liu Z-Q, Lin S, Baker PJ, et al. Transcriptome sequencing and analysis of the entomopathogenic fungus Hirsutella sinensis isolated from Ophiocordyceps sinensis. BMC Genomics. 2015;16:106.
- Lin S, Liu Z-Q, Xue Y-P, et al. Biosynthetic pathway analysis for improving the cordycepin and cordycepic acid production in Hirsutella sinensis. Appl Biochem Biotechnol. 2016;179(4):633–649.
- Ohta Y, Lee J-B, Hayashi K, et al. In vivo anti-influenza virus activity of an immunomodulatory acidic polysaccharide isolated from Cordyceps militaris grown on germinated soybeans. J Agric Food Chem. 2007;55(25):10194–10199.
- Choi JN, Kim J, Lee MY, et al. Metabolomics revealed novel isoflavones and optimal cultivation time of Cordyceps militaris fermentation. J Agric Food Chem. 2010;58(7):4258–4267.
- Oh JY, Choi W-S, Lee CH, et al. The ethyl acetate extract of Cordyceps militaris inhibits IgE-mediated allergic responses in mast cells and passive cutaneous anaphylaxis reaction in mice. J Ethnopharmacol. 2011;135(2):422–429.
- Park DK, Park H-J. Ethanol extract of Cordyceps militaris grown on germinated soybeans attenuates dextran-sodium-sulfate-(DSS-) induced colitis by suppressing the expression of matrix metalloproteinases and inflammatory mediators. Biomed Res Int. 2013;2013:10928.
- Assefa AT, Vandesompele J, Thas O. On the utility of RNA sample pooling to optimize cost and statistical power in RNA sequencing experiments. BMC Genomics. 2020;21(1):312.
- Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360.
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842.
- Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
- Nueda MJ, Tarazona S, Conesa A. Next maSigPro: updating maSigPro bioconductor package for RNA-seq time series. Bioinformatics. 2014;30(18):2598–2602.
- Suparmin A, Kato T, Dohra H, et al. Insight into cordycepin biosynthesis of Cordyceps militaris: comparison between a liquid surface culture and a submerged culture through transcriptomic analysis. PLOS One. 2017;12(11):e0187052.
- Ko KP. Isoflavones: Chemistry, analysis, functions and effects on health and cancer. Asian Pac J Cancer Prev. 2014;15(17):7001–7010.
- Choi EJ, Kim GH. The antioxidant activity of daidzein metabolites, O-desmethylangolensin and equol, in HepG2 cells. Mol Med Rep. 2014;9(1):328–332.
- Ververidis F, Trantas E, Douglas C, et al. Biotechnology of flavonoids and other phenylpropanoid-derived natural products. Part I: chemical diversity, impacts on plant biology and human health. Biotechnol J. 2007;2(10):1214–1234.
- Lee J, Doo EH, Kwon DY, et al. Functionalization of isoflavones with enzymes. Food Sci Biotechnol. 2008;17:228–233.
- Guadamuro L, Florez AB, Alegria A, et al. Characterization of four β-glucosidases acting on isoflavone-glycosides from Bifidobacterium pseudocatenulatum IPLA 36007. Food Res Int. 2017;100(Pt 1):522–528.
- Kim DH, Kim BG, Lee Y, et al. Regiospecific methylation of naringenin to ponciretin by soybean O-methyltransferase expressed in Escherichia coli. J Biotechnol. 2005;119(2):155–162.