
Abstract
The present study aims to analyse phylogenetic relationships, using internal transcribed spacer sequence data of ribosomal DNA (rDNA), across 24 Citrus species and close relatives by the evaluation of several parameters such as nucleotide substitution (r), nucleotide diversity (π) and the estimated values of transition/transversion bias (R). The observed results indicated the presence of a wide divergence pattern of rDNA in subfamily Aurantioideae. Maximum parsimony (MP) analysis inferred divergence pattern in the Citrus genus. We observed seven strongly supported clades among the subfamily Aurantioideae. We postulate that the present investigation provides a more robust topology of Citrus and its close relatives, which can significantly prove as an additional support to resolve the phylogenetic relationships in Citrus genera. Therefore, sequences of noncoding regions should exhibit more phylogenetically informative sites than the coding regions do, which is in accordance with the present study.
Abbreviations
| CAPS: | = | cleaved amplified polymorphic sequence |
| ISSR: | = | inter-simple sequence repeat |
| ITS: | = | internal transcribed spacer |
| K1: | = | transition/transversion rate for purine |
| K2: | = | transition/transversion rate for pyrimidine |
| MCL: | = | maximum composite likelihood |
| MEGA 5: | = | Molecular Evolutionary Genetics Analysis version 5 |
| MP: | = | maximum parsimony |
| NCCB: | = | National Center of Citrus Breeding |
| ORF: | = | open reading frame |
| r: | = | nucleotide substitution |
| R: | = | estimated values of transition/transversion bias |
| rDNA: | = | ribosomal DNA |
| SNP: | = | single nucleotide polymorphism |
| π: | = | nucleotide diversity |
| SRAP: | = | sequence-related amplified polymorphism |
| UV: | = | ultraviolet |
Introduction
Aurantioideae, a subfamily of family Rutaceae, presents a vast variety of commercially important genera such as Citrus and Fortunella. Interestingly, the taxonomy of Citrus is complex and still the precise number of natural species is unclear, mainly because of the sexually compatible relatives.[Citation1,2] Barrett and Rhodes [Citation3] performed a numerical taxonomy and recommended that there were only three true species within the cultivated Citrus viz. Citron (Citrus medica L.), Mandarin (Citrus reticulata Blanco) and Pummelo (Citrus grandis L. Osbeck). The origins of other species are a result of hybridization of these true species. In view of this, taxonomic characterization is critically important for the Citrus genus, which has the widest divergence reported among the fruit species and it is imperative to resolve the phylogeny in order to have a better understanding of the complexity of the genus and to develop resources for the proper sustainable development of this genus.
Several earlier attempts have been made to revisit the intra- and inter-species relationships in Aurantioideae,[Citation4–8] which have been previously constrained by restricted taxon representation, using a few inferred sequences such as restriction fragment length polymorphisms (RFLPs), or usage of traditional genetic markers such as isozymes, inter simple sequence repeats (ISSRs) or Randomly Amplified Polymorphic DNA markers (RAPDs) for phylogenetic analysis. Although previous reports exploited the sequence-based approach, these approaches were focused on higher taxonomic levels such as order and family.[Citation9–11] Morpho-taxonomy evaluation, however, has serious limitations in a complex genus like Citrus. In Citrus, molecular phylogeny at various taxonomic levels has been observed in several earlier studies through application of a wide variety of molecular markers such as SSR,[Citation12] ISSR,[Citation13] SRAP and CAPS-SNP,[Citation14] as well as using chloroplast DNA and rDNA markers.[Citation15,16]
Ribosomal DNA (rDNA) as a source of higher genetic variability has been studied extensively for classification and identification at the generic and infra generic levels in plants.[Citation17,18] Consequently, it has been successfully applied to resolve phylogeny in several models and non-model plant species such as in Triticum,[Citation19] Solanum lycopersicum,[Citation20] Oryza sativa,[Citation21] and closely related species of Citrus.[Citation15,Citation17,Citation22] In the present study, we used a comparative as well as a combined approach using several parameters such as nucleotide frequency, nucleotide substitution (r), nucleotide diversity (π) and the estimated values of transition/transversion bias (R) to provide better and significant understanding of the genetic diversity and phylogenetic relations across 24 studied Citrus species and other species related to the genus. The present investigation provides an additional support for resolving the phylogeny of the complex genus Citrus.
Materials and methods
Plant materials and genomic DNA isolation (gDNA)
Twenty-four genotypes belonging to the genus Citrus and species related to it, which includes the following major groups as listed in , were sampled from the National Center of Citrus Breeding (NCCB), Huazhong Agricultural University (HZAU), Wuhan, China. Genomic DNA of Citrus cultivars () was extracted from fresh leaves following the procedure as previously described elsewhere.[Citation23] The quality and concentration of the DNA samples were checked using a UV-1601 spectrophotometer (Shimadzu, Japan) and a sub-aliquot of the DNA was subsequently diluted to 50 ng/μL for further downstream polymerase chain reaction (PCR) analysis of internal transcribed spacer (ITS) sequences. Both the stock and diluted portions were stored at –20 °C.
Table 1. Accession list of Citrus and its related species sequenced in this study, BLASTX hits against the GenBank database, similarity score and GenBank accession numbers.
PCR amplification and sequencing of the ITS region
In our present research, the entire ITS region (including ITS-1 and ITS-2 of nuclear rDNA and the 5.8S rRNA gene) of rDNA was amplified using the primers ITS1 (5′TCCGTAGGTGAACCTGCGG3′) and (5′TCCTCCGCTTATTGATATGC3′) ITS-4 as previously described.[Citation24] Briefly, each PCR cocktail of 25 μL contained 50 ng of genomic DNA, 0.5 pmol of each primer, 0.2 mmol/L dNTPs, 1 U Taq DNA polymerase (Fermentas, Shenzhen, China), 2.5 μL of 10 times PCR buffer supplied by the manufacturer and about 2.5 mmol/L MgCl2. The amplification programme consisted of an initial denaturation step at 94 ºC for 4 min, followed by 35 cycles of 94 ºC for 45 s, 55 ºC for 60 s, 72 ºC for 90 s and a final incubation step of 72 ºC for 7 min. The PCR products obtained were further resolved by electrophoresing 10 μL of the amplified aliquot in a 1.5% agarose gel and were subsequently stained using ethidium bromide and visualized under ultraviolet (UV) light. The PCR fragments were excised and purified from the gel, using an E.Z.N.A® Gel Extraction Kit (Omega Bio-Tek, Inc., Norcross, USA) and were subsequently ligated to a pMD18-T Easy vector as per the manufacturer instructions (TaKaRa, Tokyo, Japan). The ligation product was transformed into E. coli DH-5α-competent cells, using ampicillin as a selection marker. Three positive colonies from each clone were selected and sequenced by the Uni-Gene Company (Shanghai, China).
Sequence editing, alignment and phylogenetic inference
Sequencing chromatograms obtained were analysed and vector sequences were trimmed. In all the sequenced ITS regions, after vector trimming open reading frames (ORFs) were predicted using the National Center for Biotechnology Information (NCBI) Open Reading Frame Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf). The trimmed sequences were further aligned using MUSCLE v3.70+ fix1-2 [Citation25] and the resulting alignments were manually checked. Gaps were retained for further analysis. Nucleotide diversity (π), estimated values of transition/transversion bias (R), nucleotide substitutions (r) for each nucleotide pair and cluster analysis among the 24 Citrus genotypes were estimated using MEGA 5.[Citation26] We further computed the maximum composite likelihood (MCL) estimate of the pattern of nucleotide substitution.[Citation26] For the phylogenetic inference, maximum parsimony (MP) trees were computed using MEGA 5.[Citation26] The bootstrap consensus tree inferred from 500 replicates was taken to represent the evolutionary history of the Citrus genus and its related species. In brief, the MP tree was obtained using the Subtree–Pruning–Regrafting (SPR) algorithm with search level 1 in which the initial trees were obtained with the random addition of sequences (10 replicates). The analysis involved 25 nucleotide sequences corresponding to a total of 943 positions in the final dataset. This approach has been previously followed for resolving the phylogeny of Indian Citrus cultivars.[Citation17] The phylogenetic tree was re-rooted using the ITS sequence of Atlantia monophylla (NCBI accession number, GQ225867) as reported in a previous study.[Citation17]
Results and discussion
In our study, first after subsequent cleaning of the ITS sequence, we performed homology searches against the NCBI GenBank (https://www.ncbi.nlm.nih.gov/) database, using BLASTn, which revealed 93%–100% similarity with the previously sequenced ITS regions, providing an evidence for the good trustworthiness of ITS regions sequenced in this study. All the ITS sequences of Citrus and its relative species have been submitted to GenBank databases (www.ncbi.nlm.nih.gov) and can be accessed under accession numbers as referred in . In our investigation, the universal primers ITS1 (forward) and ITS4 (reverse) amplified the complete ITS region (ITS1, 5.8S rRNA gene and ITS2) but variation for the ITS regions (650–750 bp) was observed for the individual species; however, the observed length was found similar to the ITS length variation as observed in recently sequenced ITS regions in Citrus cultivars [Citation17] and also similar to the large-scale ITS sequences in Brassicacea.[Citation27]
Recently, length variation across ITS regions has also been observed in Cymbidium species.[Citation28] The nucleotide composition showed an average of guanine and cytosine (GC) (58.5%) and AT (41.5%) content. The highest number of nucleotides for the ITS sequence was observed in C. reticulata × C. sinensis (788 bases), whereas the lowest one was recorded in C. sinensis cv. Newhall (609 bases). The maximum GC content (64%) and the lowest AT content (35.8%) were observed in the case of C. sinensis cv. Anliu, C. paradisi Macf. cv. Red Marsh grapefruit, Fortunella hindsii Swing. cv. Hongkong Kumquat, C. sinensis cv. Valencia and C. reticulata × C. sinensis (L.) Osbeck cv. Murcott (hybrid). Nevertheless, the lowest GC content (46%) and maximum AT content (54%) were recorded in Poncirus trifoliata (L) Raf. A similar GC content was observed in the sequenced ITS region of Indian cultivars, which supports the observed pattern of GC variation.[Citation17] In view of the relatively rapid evolution rate, differences in sequence and/or length of ITS rDNA are possible between close species.[Citation22] Sequence length variation in ITS and significant difference in the nucleotide composition were also observed in Cymbidium species,[Citation28] which supports our present results.
We further evaluated the nucleotide diversity value (π), using the Tajima Neutrality test.[Citation29] We observed a total of 334 segregating sites (S), 390 maximum number of positions (N) and 24 sites (M) demonstrating a higher nucleotide diversity rate (0.41) among the Citrus genus and its closely related species. It is a well-known fact that during DNA sequence evolution, the rate of transitional changes differs quite relatively from the rate of transversional changes, with transitions generally occurring more frequently than transversions. The transition/transversion bias (R) across the combined data was evaluated using Kimura two-parameter analysis with four models (K2+G+I, K2+I, K2+G and K2) to describe the best substitution pattern (). The highest number of substitutions (r) for each nucleotide pair was recorded among r (CG ±0.189), revealing high levels of substitutions. However, moderate and lower values of substitution were observed for r (AG; TC; CT; GA ±0.132) and (AC; TA; TG; CA; GT; GC ±0.059), respectively.[Citation30] The transition/transversion rate ratios observed in our analyses were K1 = 2.136 and K2 = 1.716. However, we observed a higher transition/transversion rate for purine (K1 = 2.136) as compared to recent reports in Indian cultivars (K1 = 1.716) and as compared to the transition/transversion rate for pyrimidine (K2 = 2.796).[Citation17] In our analyses, we observed that the overall transition/transversion bias is R = 0.956, which gives a strong support for the dominance of the transitions over transversion in Citrus germplasm. The observed higher transition/transversion (R) rate is in accordance with the recent reports of the observed higher transition/transversion bias (1.158) in the phylogeny in Indian Citrus cultivars recently inferred using ITS.[Citation17] The present rate of transition/transversion bias is also in complete agreement with a recently observed transition bias in Citrus germplasm using SSR markers.[Citation1]
Table 2. Maximum-likelihood fits, using the Kimura two-parameter model among 24 different nucleotide sequences for combined data of ITS.
Up to now, there are only a few reports, which sufficiently explain the significance of ITS rDNA as a molecular genotyping tool in Citrus.[Citation2,Citation17,Citation22] We analysed the evolutionary history of Citrus and its relative species by the MP method, using the SPR algorithm with search level 1 in which the initial trees were obtained by the random addition of sequences (10 replicates) as implemented in MEGA5.[Citation26] The analysis involved 25 nucleotide sequences and there were a total of 943 positions in the final dataset for MP analysis. In the present investigation, using our data, we obtained the most parsimonious tree with a length of 1818 (). MP inferred a consistency index (CI – 0.666868), a retention index (RI – 0.792247) and a composite index of 0.551697. The phylogenetic tree was re-rooted using A. monophylla (NCBI accession number, GQ225867) as an outgroup species (). Recently, A. monophylla has been used as an outgroup for inferring the phylogeny in Citrus cultivars.[Citation17] In a recent study, using the ITS sequence data and MP analysis, a similar consistency (0.6804) and retention (0.7350) index was observed, which is in line with our observed results and supports the present MP phylogenetic inference.[Citation17]
Figure 1. Maximum parsimony analysis of genotypes of the Citrus genus and its related species based on ITS rDNA data.
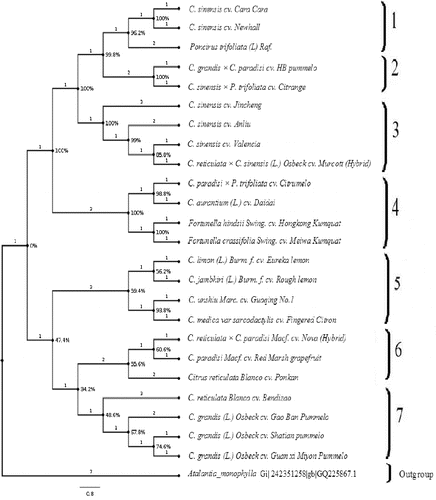
The phylogenetic analysis revealed several well-supported clades with strong bootstrap values. In total, we observed seven strongly supported clades, which were clearly distinguishable among the subfamily Aurantioideae. The first clade was clustered jointly C. sinensis cv. Cara Cara, C. sinensis cv. Newhall (navel oranges) and Poncirus trifoliata with a bootstrap value of 96.2%. In context, the second clade represents the C. grandis × C. paradisi cv. HB pummelo and C. sinensis × P. trifoliata cv. Citrange. Additionally, C. sinensis cv. Jincheng, C. sinensis cv. Anliu, C. sinensis cv. Valencia and C. reticulata × C. sinensis L. Osbeck cv. Murcott (hybrid) were placed together in the third clade with bootstrap values of 100%. The phylogenetic analysis, as inferred for the ITS sequence data indicated that sweet oranges (Jincheng, Anliu and Valencia) showed a close relationship with the hybrid Murcott (C. reticulata × C. sinensis). The observed clades are in strong support with the previously observed clades.[Citation2,Citation5,Citation15,16,Citation31,32]
The genus Fortunella contains the Kumquats. It closely resembles Citrus species, although their morphology is very different. Across the fourth clade, C. paradisi × P. trifoliata cv. Citrumelo, C. aurantium (L.) cv. Daidai, Fortunella hindsii Swing. cv. Hongkong Kumquat and Fortunella crassifolia Swing. cv. Meiwa Kumquat were grouped in a sister clade. Morphologically, Fortunella and Citrus are significantly different from each other. However, there are previous evidences of nested clustering of Fortunella with Citrus species.[Citation8,Citation12] Kyndt et al. [Citation2] demonstrated the inclusion of the Fortunella spp. within Citrus, close to the C. reticulata group, confirming their recent reclassification as C. japonica, using ITS sequence polymorphism. Our ITS rDNA data showed a close evolutionary relationship between Fortunella (Hongkong and Meiwa Kumquat) and sour orange (C. aurantium). Biswas et al. [Citation33] reported that Fortunella might be less divergent than Citrus at the molecular level than observed in morphology. Moreover, C. medica var sarcodactylis cv. Fingered Citron, C. unshiu Marc. cv. Guoqing No.1, C. jambhiri (L.) Burm. f. cv. Rough lemon and C. limon (L.) Burm. f. cv. Eureka lemon occupies the fifth clade. Several earlier experts hypothesized C. limon to have a complex hybrid origin of Citron and Lime [Citation34–36] or Citron and Sour Orange [Citation7,Citation37] or Sour Orange and Lime.[Citation38,39] In our study, C. limon was grouped with C. jambhiri and C. medica var sarcodactylis in the MP tree, which supports the close relationship among these species. In a previous study by Jena et al.,[Citation15] they proposed a close relationship between C. jambhiri and C. reticulata and supported the role of C. reticulata as a maternal parent in the hybrid origin of C. jambhiri, which perfectly fits with the phylogenetically observed clade in our analyses.
The citron mitotype contained only C. medica. This species did not transmit its cytoplasm to other species but played an important role as a male parent.[Citation32] Indeed, our results confirmed that citron was grouped with C. jambhiri (L.) Burm. f. cv. Rough lemon and C. limon (L.) Burm. f. cv. Eureka Lemon. Furthermore, Citrus reticulata Blanco cv. Ponkan, C. paradisi Macf. cv. Red Marsh grapefruit and C. reticulata × C. paradisi Macf. cv. Nova (hybrid) were grouped jointly as a sixth clade. As it is well known, the origin of grapefruit has been well documented and is considered to have originated most probably from a hybrid between pummelo and sweet orange, perhaps through back introgression to pummelo.[Citation5,Citation40] In our study, the ITS rDNA data indicated that C. paradisi Macf. cv. Red Marsh grapefruit and the C. reticulata × C. paradisi Macf. cv. Nova (hybrid) were grouped together with pummelo, supporting the viewpoint of a backcross with pummelo.[Citation8] For C. grandis, C. grandis L. Osbeck cv. Guan xi Miyon Pummelo, C. grandis L. Osbeck cv. Gao Ban pummelo and C. grandis L. Osbeck cv. Shatian pummelo were grouped with C. reticulata Blanco cv. Bendizao in the last clade. In addition, Shatian pummelo, Gao Ban pummelo and Guan xi Miyon Pummelo (C. grandis) were clustered with C. reticulata (Ponkan mandarin). In contrast, HB pummelo (C. grandis × C. paradisi) is closer to Citrange (C. sinensis × P. trifoliata). It is generally accepted that citrons, mandarin and pummelo are three true species in the genus Citrus.[Citation33,Citation40] Our data inferred a close genetic relationship between mandarin and pummelo, concordant with the previous results of Xu et al.,[Citation22] which supports this theory, as mandarin and pummelo each had a near uniform ITS rDNA sequence. In this context, Froelicher et al. [Citation32] strongly proposed that mandarin played an important role in the evolution of cultivated Citrus; in addition to this, the authors supported that pummelo mitotype was found to be present as the most important cultivated Citrus species. Furthermore, Barkley et al. [Citation12] reported that it was a mixture between the citron, mandarin and pummelo groups with the majority of its alleles coming from the citron and mandarin groups.
Conclusions
To conclude, the present study presents an effective utilization of rDNA sequence divergence to maximize the possible knowledge of the genetic diversity within the Citrus genus and its relatives. The phylogenetic tree of the rDNA supported seven strong clades which were clearly shown among the genus Citrus. Consequently, this study not only corroborated the previous molecular reconstruction of subfamily Aurantioideae, but also strengthened previous claims concerning the evolutionary biology of the genus Citrus.
Acknowledgements
Mohamed Amar thanks Huazhong Agricultural University for providing post-doctoral fellowship. We are grateful to Prof. F. M. Abdel-Tawab, Dr Wen-Fang Zeng and Mr Xin-Jian Zhang for their constructive comments. Special thanks are given to Bioversity International for their valuable support.
Additional information
Funding
References
- Garcia-Lor A, Curk F, Snoussi-Trifa H, Morillon R, Ancillo G, Luro F, Navarro L, Ollitrault P. Ann Bot. 2013;111:1–19. Available from: http://dx.doi.org/10.1093/aob/mcs227
- Kyndt T, Dung TN, Goetghebeur P, Toan HT, Gheysen G. Genet Resour Crop Evol. 2010;57:183–192. Available from: http://dx.doi.org/0.1007/s10722-009-9460-0
- Barrett HC, Rhodes AM. Syst Bot. 1976;1:105–136. Available from: http://dx.doi.org/10.2307/2418763
- Abkenar AA, Isshiki S, Tashiro Y. Sci Hortic. 2004;102:233–242. Available from: http://dx.doi.org/10.1016/j.scienta.2004.01.003
- Bayer RJ, Mabberley DJ, Morton C, Miller CH, Sharma IK, Pfeil BE, Rich S, Hitchcock R, Sykes S. Am J Bot. 2009;96:668–685. Available from: http://dx.doi.org/10.3732/ajb.0800341
- Fang DQ, Krueger RR, Roose ML. J Am Soc Hortic Sci. 1998;123:612–617.
- Nicolosi E, Deng ZN, Genetile A, La Malfa S, Continella G, Tribulato E. Theor Appl Genet. 2000;100:1155–1166. Available from: http://dx.doi.org/10.1007/s001220051419
- Pang XM, Hu CG, Deng XX. Genet Resour Crop Evol. 2007;54:429–436. Available from: http://dx.doi.org/10.1007/s10722-006-0005-5
- Chase MW, Morton CM, Kallunki JA. Am J Bot. 1999;86:1191–1199. Available from: http://dx.doi.org/10.2307/2656983
- Groppo M, Pirani JR, Salatino ML, Blanco SR, Kallunki JA. Am J Bot. 2008;95:985–1005. Available from: http://dx.doi.org/10.3732/ajb.2007313
- Poon WS, Shaw PC, Simmons MP, But PPH. Syst Bot. 2007;32:837–846. Available from: http://dx.doi.org/10.1600/036364407783390692
- Barkley NA, Roose ML, Krueger RR, Federici CT. Theor Appl Genet. 2006;112:1519–1531. Available from: http://dx.doi.org/10.1007/s00122-006-0255-9
- Shahsavar AR, Izadpanahm K, Tafazoli E, Tabatabaeic BES. Sci Hortic. 2007;112:310–314. Available from: http://dx.doi.org/10.1016/j.scienta.2006.12.039
- Amar MH, Biswas MK, Zhang Z, Guo WW. Sci Hortic. 2011;128:220–227. Available from: http://dx.doi.org/10.1016/j.scienta.2011.01.021
- Jena SN, Kumar S, Nair NK. Sci Hortic. 2009;119:403–416. Available from: http://dx.doi.org/10.1016/j.scienta.2008.08.030
- Lu Z, Zhou Z, Xie R. Agric Sci China. 2011;10:49–57. Available from: http://dx.doi.org/10.1016/S1671-2927(11)60306-4
- Kumar S, Nair KN, Jena SN. Genet Resour Crop Evol. 2013;60:59–75. Available from: http://dx.doi.org/10.1007/s10722-012-9814-x
- Salares R, Brown TA. Genet Resour Crop Evol. 2004;51:701–712. Available from: http://dx.doi.org/10.1023/B:GRES.0000034576.34036.a1
- Qian J, Sun Y, Duan Y. Acta Agro Sinica. 2009;35:1021–1029. Available from: http://dx.doi.org/10.1016/S1875-2780(08)60088-7
- Jo SH, Koo DH, Kim JF, Hur CG, Lee S, Yang TJ, Kwon SY, Choi D. BMC Genet. 2009;9:42. Available from: http://dx.doi.org/10.1186/1471-2229-9-42
- Chang KD, Fang SA, Chang FC, Chung MC. Genomics. 2010;96:181–190. Available from: http://dx.doi.org/10.1016/j.ygeno.2010.05.005
- Xu CJ, Bao L, Zhang B, Bei ZM, Ye XY, Zhang SL, Chen KS. Plant Breeding. 2006;125:519–522. Available from: http://dx.doi.org/10.1111/j.1439-0523.2006.01263.x
- Cheng YJ, Guo WW, Yi HL, Pang XM, Deng XX. Plant Mol Biol Rep. 2003;21:177–178. Available from: http://dx.doi.org/10.1007/BF02774246
- White TJ, Bruns TD, Lee SB, Taylor JW. PCR protocols: a guide to methods and applications. San Diego (CA): Academic Press; 1990. p. 315–322.
- Edgar RC. Nucleic Acids Res. 2004;32:1792–1797. Available from: http://dx.doi.org/10.1093/nar/gkh340
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. Mol Biol Evol. 2011;28:2731–2739. Available from: http://dx.doi.org/10.1093/molbev/msr121
- Warwick SI, Mummenhoff K, Sauder CA, Koch MA, Al-Shehbaz IA. Plant Syst Evol. 2010;285:209–232. Available from: http://dx.doi.org/10.1007/s00606-010-0271-8
- Sharma SK, Dkhar J, Kumaria S, Tandon P, Rao SR. Gene. 2012;495:10–15. Available from: http://dx.doi.org/10.1016/j.gene.2011.12.052
- Tajima F. Genetics. 1989;123:585–595.
- Tamura K, Nei M. Mol Biol Evol. 1993;10:512–526.
- Chibame KM, Laskar MA. Sci Hortic. 2010;124:345–348. Available from: http://dx.doi.org/10.1016/j.scienta.2010.01.014
- Froelicher Y, Mouhaya W, Bassene JB, Costantino G, Kamiri M, Luro F, Morillon R, Ollitrault P. Tree Genet Genomes. 2011;7:49–61. Available from: http://dx.doi.org/10.1007/s11295-010-0314-x
- Biswas MK, Chai LJ, Amar MH, Zhang XL, Deng XX. Sci Hortic. 2011;129:798–803. Available from: http://dx.doi.org/10.1016/j.scienta.2011.06.015
- Malik MN, Scora RW, Soost RK. Hilgardia. 1974;42:361–382.
- Scora RW. Bull Torrey Bot Club. 1975;102:369–375. Available from: http://dx.doi.org/10.2307/2484763
- Swingle WT. The Citrus industry. Vol. 1, Berkeley: University of California; 1943. p. 128–474.
- Gulsen O, Roose ML. J Am Soc Hortic Sci. 2001;126:309–317.
- Hirai M, Kozaki I. Proc Int Soc Citricult. 1981;1:1–13.
- Torres AM, Soost RK, Diedenhofen U. Am J Bot. 1978;65:869–881. Available from: http://dx.doi.org/10.2307/2442183
- Moore GA. Trends Genet. 2001;17:536–540. Available from: http://dx.doi.org/10.1016/S0168-9525(01)02442-8