
Abstract
Highly specific, sensitive and rapid tests are required for the detection and identification of covert bacterial contaminations in plant tissue cultures. Current methods available for this purpose are tedious, time consuming, highly error prone, expensive, require advanced technical expertise and are sometimes ineffective. We report here the development of a sensitive polymerase chain reaction (PCR) based method for the rapid detection and identification of bacteria occurring in plant tissue cultures. A total of 121 16S ribosomal DNA (rDNA) coding regions from 14 different groups of bacteria, algae and plants, available in the Gene Bank/European Molecular Biology Laboratory databases, were aligned and several conserved DNA sequences of bacterial origin were identified. From those, five degenerated primers were designed in order to amplify only the bacterial DNA present in mixed plant/bacteria genomic DNA extracts. A known amount of bacterial suspension of either covert Pseudomonas or covert Bacillus were added to in vitro plant leaves and total plant/bacterial DNA extracted using three different methods to determine the lowest number of bacteria required to be present in order to allow their detection. The highest sensitivity of the bacterial cell detection was 2.5 × 106 cells of both Bacillus and Pseudomonas inoculums, using template DNA prepared by the MiniPrep method. Generation of PCR amplification fragments was achieved only for the 16S rDNA bacterial gene by using four combinations of degenerated primers. Successive sequence analysis of these amplified fragments led to the rapid detection and molecular identification of bacteria covertly associated with plants.
Introduction
Plant cell and tissue culture is an important tool for the aseptic production of cells, tissues and organs in both basic and applied research. It has been extensively employed in commercial production of plant metabolites, biotransformation of pharmaceuticals, production of proteins including antibiotics, plant genetic manipulation and massive production of plants in the horticultural industry.[Citation1] It is very important that in vitro cultures are free of biological contamination and are maintained as aseptic cultures during manipulation, growth and storage. Contamination management is focused on the elimination of micro-arthropods, fungi, bacteria and viruses/viroids.[Citation2] The danger of contaminants is well known in the majority of commercial and scientific plant tissue culture laboratories and losses due to contamination still average between 3% and 15% at every subculture. Most of these constantly occurring losses are caused by fungal, yeast, and bacterial contaminants.[Citation3] Roughly 20%–55% of contamination losses to in vitro plant cultures are caused by bacteria.[Citation4] Furthermore, loss of valuable research material in micropropagation is frequently caused by endophytic pathogenic bacteria and viruses. Despite the awareness of the contamination problem, many laboratories are still troubled by sudden outbreaks of bacterial infestations resulting in the loss of culture stocks that had been free of visible contamination for long periods of time.[Citation2]
Identification of microorganisms by conventional laboratory tests takes only a few days and that by molecular approaches reduces the identification time to hours. The emergence of DNA diagnostics is revolutionizing the whole approach in identifying and monitoring microbes. Such methods use genotypic rather than phenotypic markers to identify specific microbes and the strengths of DNA diagnostics lie in the fact that: (1) nucleic acids can be rapidly and sensitively measured and (2) the sequence of nucleotides in a given DNA molecule is sufficiently specific to be used for reliable diagnosis. Similar diagnostic studies have been exclusively and specifically developed for anammox bacteria in soils [Citation5] and for identifying the genetic diversity of 60 Methylobacterium spp. strains from eight different host plants.[Citation6] Both diagnostic studies target the 16S ribosomal gene of bacteria. Identification of bacteria isolated from in vitro cultures of Billbergia magnifica ssp. acutisepalia revealed many important species such as Bacillus cereus, Bacillus thuringiensis, Bacillus fusiformis, Agrobacterium sp., Microbacterium sp., Sphingomonas sp., Pseudomonas putida and Paenibacillus amylolyticus, which were identified by sequencing of the 16S ribosomal DNA (rDNA) gene.[Citation7]
In this study, we describe a polymerase chain reaction (PCR) based strategy and associated protocols that allow the rapid detection of two bacterial contaminants in tissue-cultured plants of the ornamental plant Billbergia by amplifying only a fragment of the 16S rDNA gene of the bacteria and not the conserved region of the 16S ribosomal gene of cpDNA. This PCR product can be subsequently used for classification of the detected bacteria by cloning and sequencing.[Citation8]
Materials and methods
Alignment of 16S rRNA sequences of different organisms
In order to identify conserved regions of the 16S rDNA gene in bacteria and the prokaryotic DNA of plants, DNA sequences of the 16S region belonging to (1) 79 bacteria of known phyla, (2) 10 uncultured bacteria, (3) 10 algae and (4) 22 plants () were aligned using the ClustalX from the DNAStar Nucleotide Sequence Analysis Package. The 16S rDNA sequences were obtained from the National Center for Biotechnology Information (NCBI) database.
Table 1. List of organisms of the 16S rDNA sequences aligned and accession numbers.
Design of oligonucleotide primers
To amplify only fragments of bacterial 16S rDNA found in plant tissues, five degenerated oligonucleotide primers were designed (and produced upon instructions by Life Technologies, UK) in the conserved regions of the 16S rDNA gene, based on the sequences of 121 different bacteria and algae/plant chloroplast DNA (), in order to be universal for most bacteria ( and ).
Table 2. Combinations of primers that detect bacteria in plant tissues of Billbergia magnifica ssp. acutisepalia.
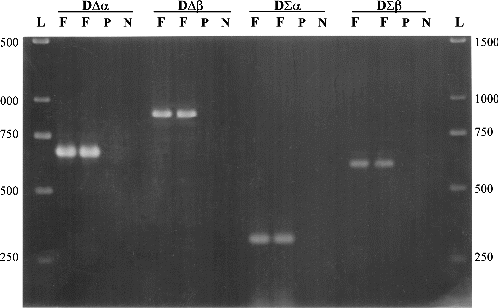
Figure 1. Detection of bacterial DNA in plant tissues, using four sets of primers. F is an amplified fragment (in duplicated reactions) at different positions of the bacterial 16S rDNA gene. P is the control template using pure plant DNA and N is the negative control with no DNA template. L is 1 kb ladder (GeneRulerTM, MBI Fermentas, Lithuania).
Origin of bacterial isolates and plant tissue
Bacterial isolates used for these experiments were a Gram-positive Bacillus sp. and a Gram-negative Pseudomonas sp. obtained in previous studies [Citation9] and which could be classified by sequencing of their 16S rDNA gene. The plant tissue used for DNA extraction was derived from microbe-free clonal in vitro plants of B. magnifica ssp. acutisepalia produced by regeneration of plants via somatic embryogenesis.[Citation7] The latter plants did not yield any detectable and culturable microbes following extensive and rigorous enrichment plating tests.[Citation9]
Estimation of the number of bacterial cells
Three different methods were used to estimate the number of bacterial cells present per inoculum. Ten-millilitre aliquots of Trypto-Soya Broth medium (Oxoid, Unipath Ltd) were inoculated with single bacterial colonies and incubated overnight in an orbital shaker (Orbital mixer, Denley Ltd, UK) at 200 r/min under room temperatures (25 ± 3 °C). Bacterial cells were washed twice with 10 mmol/L MgCl2 and were resuspended in the same buffer; then, the absorbance of the suspensions was adjusted to an optical density of 1 (at 600 nm), using a spectrophotometer (SP8-100 UV/VIS, Pye/ATI Unicam/Philips, Spectronic Camspec Ltd, UK).
To estimate the number of bacterial cells in each suspension, three 100 μL aliquots were plated onto Trypto-Soya Agar (TSA) media [30 mL on each (9 cm) Petri dish (Sterilin, UK)] using a sterile glass loop to spread each aliquot evenly over the media. The plates were incubated at 25 °C for 24 h and single colonies counted in order to estimate the number of bacterial cells in the suspension. Estimation of the bacterial cells was also made by the counting chamber method (Weber Scientific International Ltd, England). A drop of suspension, with an estimated OD600 of 0.1, was placed on the slide, and bacterial cells were counted under a dissecting microscope (Dialux 20 EB, Ernst Leitz Wetzlar GMBH, Germany). Following this standardization, 10-fold dilutions were then derived ().
Table 3. Estimation of bacterial cell densities used as inoculums.
Sample preparation prior to DNA extraction
Samples of 50, 250 and 500 mg of plant tissue were weighed on a digital top-pan balance (Sartorius, Fisher Ltd, UK) and placed into 1.5 mL sterile microtubes. A total of 50 μL of bacterial inoculum was added to each microtube after the plant tissue had been homogenized in the buffer being tested using a plastic sterile pestle. Bacterial inoculums were prepared in serial 10-fold dilutions (α1, β1, γ1) for Gram-positive and (α2, β2, γ2) for Gram-negative bacteria, after estimating the number of bacteria present in the stock suspension using 10 mmol/L MgCl2.
Nucleic acid preparation
Three different DNA extraction methods were tested for their capacity to extract nucleic acids from bacteria/plant mixtures, from pure cultures of bacteria and from non-contaminated in vitro plant tissue. The latter samples were used also as negative controls for the PCR reactions.
The first protocol was the MiniPrep method [Citation10] and was based on chemical disruption of cells, using mercaptoethanol following isoamyl alcohol/chloroform separation. This protocol was utilized for contaminated plant samples, which were maintained previously in a freezer; mortars and pestles were thoroughly cleaned and kept in a freezer (at least overnight at −20 °C). Contaminated plant samples were homogenized using liquid nitrogen and 150 mg of the homogenized tissues was transferred into a new 1.5 mL sterile microcentrifuge tube. The next steps involved: (1) addition of 600 μL DNA extraction buffer [3% (w/v) cetyl trimethylammonium bromide (CTAB), 1.4 mol/L NaCl, 20 mmol/L ethylenediaminetetraacetic acid (EDTA) pH 8.0, 100 mmol/L Tris–HCl pH 8.0 and 1% (w/v) polyvinylpyrrolidone PVP-40T (soluble)] and mixing well by vortexing for 30 s, (2) addition of 20 μL 2-mercaptoethanol and mixing well by vortexing for 30 s, (3) incubation of the homogenized samples at room temperature for 10 min, (4) addition of 250 μL chloroform/isoamyl ethanol, 24:1 (v/v) and mixing very well by vortexing for 30 s, (5) phase separation by microcentrifugation (at 3380g) for 15 min, (6) transfer of supernatant in a new 1.5 mL sterile microcentrifuge tube, (7) addition of 250 μL chloroform/isoamyl ethanol, 24:1 (v/v) and mixing very well by vortexing for 30 s, (8) phase separation by microcentrifugation (at 3380g) for 10 min, (9) transfer of supernatant in a new 1.5 mL sterile microcentrifuge tube and an estimate made of the total volume of supernatant, (10) addition of 2.5 volumes of 100% ethanol into the volume of supernatant (step 9), (11) mixing gently and sample incubation at room temperature for 90 min, (12) microcentrifugation (at 17,100g) for 10 minutes, (13) supernatant removal, addition of 500 μL of 70% ethanol and vortexing thoroughly for 1 min, (14) microcentrifugation (at 17,100g) for 3 min and removal of ethanol, (15) air-dry the pellet for 10 min at 37 °C, (16) resuspension of the pellet in 50–100 μL of molecular grade water or Tris–EDTA (TE) buffer, (17) storage of DNA solution at −20 °C.
The second method was the protocol of Lawson et al. [Citation11] for bacterial DNA extraction; this involved disrupting cells with an enzymatic procedure followed by phenol/chloroform separation. The third method tested was the RapidPrep® Micro Genomic DNA Isolation Kit for cells and tissue [Citation12] which involved isolating DNA by means of anion-exchange chromatography in a spin-column format.
PCR amplification
The PCR mixtures (50 μL) contained 5 μL of 10× reaction buffer [16 mmol/L (NH4)2SO4; 70 mmol/L Tris–HCl pH 8.8; 0.1% Tween 20]; 0.06 μL of each deoxynucleoside triphosphate (100 mmol/L); 0.15 μL of BioTaq (5 u/μL; Thermus aquaticus; Bioline); 1.5 μL MgCl2 (50 mmol/L); 2 μL of each degenerated primer (30 pmol/L); 1 μL template DNA (approximately 10–40 ng); and 38.11 μL of analytical grade water. PCR amplification was performed with a thermal cycler (GenAmp In Situ PCR System 1000, PerkinElmer Cetus); cycles consisted of 6 min denaturation at 96 °C followed by 40 cycles of 30 s at 95 °C for further denaturation, 15 s at 59 °C (for DΣβ) primer set annealing, 1 min at 72 °C extension and ended with a 10 min extension at 72 °C. The annealing temperature for the other set of primers is shown in .
After amplification, 6 μL of DNA loading buffer (20% glycerol, 5 mmol/L EDTA, bromophenol blue) was added and 16 μL of this mixture was separated by electrophoresis in a 1.5% agarose gel in Tris–acetate–EDTA electrophoresis buffer (pH 8.5) at room temperature. Gels were stained for 20 min in ethidium bromide (0.75 μg/L in deionized water), distained (5 min in deionized water) and viewed on a UV transilluminator.
Results and discussion
Two forward and three reverse oligonucleotide primers were designed () on the basis of a total of 121 different published 16S rDNA sequences derived from eubacteria, archae and chloroplasts of algae and plants. Using bacterial DNA from a Pseudomonas sp. isolate and plant genomic DNA from in vitro plants of Billbergia, combinations of the degenerated primers amplified four different fragments in the 16S rDNA gene of bacteria, without amplifying chloroplastic DNA (). The extent to which plant/bacteria mix lysates can be amplified is largely dependent on the method used to extract DNA. Enzymes and inhibitors in the lysate can have a significant, deleterious effect on the efficiency and sensitivity of PCR assays.[Citation13] For this reason, three different DNA extraction procedures were compared to test their effects on PCR amplification using the DΣβ pair of primers. The results of PCR efficiency and sensitivity using DNA templates from each DNA extraction method showed that the best level of detection was achievable at approximately 2.5 × 106 bacterial cells per 50 mg in vitro tissue of Billbergia, for both Gram-positive and Gram-negative bacteria (). The MiniPrep extraction method [Citation10] produced a strong PCR amplification fragment (), compared to the faint band produced by DNA extracted by the RapidPrep® method ((B)) and the absence of a band for DNA extracted by Lawson's method ((A)). The sensitivity of the MiniPrep method in different ratios of bacteria/plant tissue used for DNA extraction was examined. A total of 2.5 × 107 bacterial cells in 500 mg in vitro tissue of Billbergia were detected but the template DNA did not produce as strong an amplification product as those in which either 50 or 250 mg plant tissue was used. Generally the lesser the plant tissue used for DNA extraction, the lower the number of bacterial cells detected ().
Figure 2. Detection of bacterial DNA in plant tissues, using the DΣβ pair of primers (). Different DNA templates were tested for both Gram-positive and Gram-negative bacteria used as inoculum with either 50, 250 or 500 mg in vitro tissue of Billbergia and extracted by the MiniPrep method.[Citation10] P is the control using pure plant DNA as a template and N is the negative control with no DNA template; α1, β1, γ1 and α2, β2, γ2 are different concentrations of bacterial cells used as inoculum ().
![Figure 2. Detection of bacterial DNA in plant tissues, using the DΣβ pair of primers (Table 2). Different DNA templates were tested for both Gram-positive and Gram-negative bacteria used as inoculum with either 50, 250 or 500 mg in vitro tissue of Billbergia and extracted by the MiniPrep method.[Citation10] P is the control using pure plant DNA as a template and N is the negative control with no DNA template; α1, β1, γ1 and α2, β2, γ2 are different concentrations of bacterial cells used as inoculum (Table 3).](/cms/asset/cc3ee274-3eeb-4df6-b2db-9fea3a7b26a0/tbeq_a_937139_f0002_b.gif)
Figure 3. Detection of bacterial DNA in plant tissues, using the DΣβ pair of primers () that amplify an approximately 620 bp fragment in the 16S rDNA gene of Gram-positive and Gram-negative bacteria present in Billbergia tissues. DNA extracts used as template for PCR amplification obtained by (A) Lawson's method for extraction of bacterial genomic DNA [Citation11] and (B) RapidPrep® Micro Genomic DNA Isolation Kit for cells and tissues. P is the control using pure plant DNA as a template and N is the negative control with no DNA template; α1, β1, γ1 and α2, β2, γ2 are different concentrations of bacterial cells used as inoculum ().
![Figure 3. Detection of bacterial DNA in plant tissues, using the DΣβ pair of primers (Table 2) that amplify an approximately 620 bp fragment in the 16S rDNA gene of Gram-positive and Gram-negative bacteria present in Billbergia tissues. DNA extracts used as template for PCR amplification obtained by (A) Lawson's method for extraction of bacterial genomic DNA [Citation11] and (B) RapidPrep® Micro Genomic DNA Isolation Kit for cells and tissues. P is the control using pure plant DNA as a template and N is the negative control with no DNA template; α1, β1, γ1 and α2, β2, γ2 are different concentrations of bacterial cells used as inoculum (Table 3).](/cms/asset/4c02c71c-f43c-48ff-bf2d-6c9cb970be19/tbeq_a_937139_f0003_b.gif)
Micropropagation of the epiphytic bromeliad B. magnifica ssp. acutisepalia from vegetative explants is difficult and in many cases impossible due to heavily contaminated explant material used to initiate in vitro cultures.[Citation7] Our previous studies showed that many different contaminant bacterial species could be recovered despite the fact that harsh chemical surface sterilization treatments were used.[Citation9] In order to study the microcosmos of Billbergia and the interactions of those microbes with the plant during the micropropagation and post-weaning processes, we developed a PCR-based bacterial detection tool.
The degenerated primers designed in this study were based on the sequences of the 16S rDNA gene belonging to all bacterial phyla and most of the known bacterial families available in the Gene Bank/EMBL (European Molecular Biology Laboratory) databases. Most of the 16S rDNA gene sequences of plant chloroplasts that were available in the Gene Bank/EMBL databases showed a high degree of similarity. The sequence of the 16S rDNA gene of Billbergia was also very similar to other plants listed in the databases and the conserved regions where the degenerated primers designed were almost identical. Thus, the degenerated primers designed for Billbergia would appear to have the potential to be used for other plants as well.
The molecular detection of bacterial contaminants in plant tissue cultures indicates that: (1) specific oligonucleotide primers (at genus, family or even at phylum level) can be designed and can detect bacteria present not only in tissues of B. magnifica ssp. acutisepalia but in other plants as well; (2) primers can amplify members of culturable and unculturable bacterial and archaeal groups ( and ); (3) the isolation protocol used for the extraction of genomic DNA affects the template purity and most importantly the efficiency of the PCR reactions, as previously shown by Simon et al. [Citation14] and in the course of the present study ( and ). The generated PCR product can be subsequently used for identification of the bacteria harboured in the plant tissues, by cloning and sequencing.
Conclusions
The results of the present study showed that specific oligonucleotide primers (at genus, family or even at phylum level) can be designed and used for molecular detection of bacterial contaminants not only in tissue cultures of B. magnifica ssp. acutisepalia but in other plants as well. A set of primers that can amplify members of culturable and unculturable bacterial and archaeal groups was proposed. The isolation protocol used for the extraction of genomic DNA was observed to affect the template purity and most importantly the efficiency of the PCR reactions. The generated PCR product can be subsequently used for identification of the bacteria harboured in the plant tissues, by cloning and sequencing.
Additional information
Funding
References
- Smith RH. Plant tissue culture. 3rd ed. San Diego (CA): Academic Press; 2013.
- Cassells AC. Pathogen and biological contamination management in plant tissue culture: phytopathogens, vitro pathogens and vitro pests. In: Lojola Vargas VM, Ochoa Alejo N, editors. Plant cell culture protocols, methods in molecular biology. Vol. 877. New York (NY): Humana Press; 2012. p. 57–80.
- Boxus PH, Terzi JM. Control of accidental contamination during mass propagation. Acta Hortic. 1988;225:189–193.
- Leifert C, Morris CE, Waites WM. Ecology of microbial saprophytes and pathogens in tissue culture and field-grown plants: reasons for contamination problems in vitro. Crit Rev Plant Sci. 1994;13:139–183.
- Humbert S, Tarnawski S, Fromin N, Mallet MP, Aragno M, Zopfi J. Molecular detection of anammox bacteria in terrestrial ecosystems: distribution and diversity. ISME J. 2010;4:450–454.
- Dourado MN, Dini Andreote Fe, Dini-Andreote Fr, Conti R, Araújo JM, Araújo WL. Analysis of 16S rRNA and mxaF genes revealing insights into Methylobacterium niche-specific plant association. Genet Mol Biol. 2012;35:142–148.
- Tsoktouridis G. Molecular detection and characterisation of bacteria intimately associated with in vitro cultures of Billbergia magnifica ssp. acutisepalia during micropropagation by axillary shoot proliferation and somatic embryogenesis [dissertation]. Wye (UK): Imperial College, University of London; 2001. p. 279.
- Woese CR. Microbial evolution. Microbiol Rev. 1987;51:221–271.
- Tsoktouridis G, Mantell S, Bantinaki E, Thangavelu M. DNA profiling and strategies for in planta localisation of bacteria intimately associated with Billbergia magnifica ssp acutisepalia. Plant Cell Tissue Organ Cult. 1998;52:41–45.
- Mneney E, Mantell S, Tsoktouridis G, Amin S, Bessa A, Thangavelu M. RAPD-profiling of Tanzanian cashew (Anacardium occidentale L.). In: Topper CP, Caligari PDS, Kullaya AK, Shomari SH, Kasuga LJ, Masawe PAL, Mpunami AA, editors. Proceedings of the International Cashew and Coconut Conference: trees for life – the key to development. Reading (UK): BioHybrids International Ltd; 1998. p. 108–115.
- Lawson PA, Gharbia SE, Shah HN, Clark DR. Recognition of Fusobacterium nucleatum subgroups Fn-1, Fn-2 and Fn-3 by ribosomal RNA gene restriction patterns. FEMS Microbiol Lett. 1989;65:41–46.
- Pharmacia Biotech. Instructions for RapidPrep® Micro Genomic DNA Isolation Kit for cells and tissues. 1995.
- Leveque F, Jouan A, Deloince R, Crance M, Gratier D. Removal of PCR inhibitors. Trav Sci Chercheurs Serv Sante Armee. 1994;15:173–174.
- Simon MC, Gray DI, Cook N. DNA extraction and PCR methods for the detection of Listeria monocytogenes in cold-smoked salmon. Appl Environ Microbiol. 1996;62:822–824.