
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
In this study, time and memory optimized (TMO) algorithm is presented. Compared with Smith–Waterman's algorithm, TMO is applicable for a more accurate detection of continuous insertion/deletions (indels) in genes’ fragments, associated with disorders caused by over-repetition of a certain codon. The improvement comes from the tendency to pinpoint indels in the least preserved nucleotide pairs. All nucleotide pairs that occur less frequently are classified as less preserved and they are considered as mutated codons whose mid-nucleotides were deleted. Other benefit of the proposed algorithm is its general tendency to maximize the number of matching nucleotides included per alignment, regardless of any specific alignment metrics. Since the structure of the solution, when applying Smith–Waterman, depends on the adjustment of the alignment parameters and, therefore, an incomplete (shortened) solution may be derived, our algorithm does not reject any of the consistent matching nucleotides that can be included in the final solution. In terms of computational aspects, our algorithm runs faster than Smith–Waterman for very similar DNA and requires less memory than the most memory efficient dynamic programming algorithms. The speed up comes from the reduced number of nucleotide comparisons that have to be performed, without having to imperil the completeness of the solution. Due to the fact that four integers (16 Bytes) are required for tracking matching fragment, regardless its length, our algorithm requires less memory than Huang's algorithm.
Introduction
In order to detect mutual similarities (differences) between two or more biological sequences (DNA, RNA or protein), the sequences have to be aligned. By aligning two or more biological sequences, a model that reveals not only the elements' matching and mismatching positions, but also the positions of elements' deletions (insertions) is generated. From an applicational viewpoint, the alignment is used for phylogenetic analysis, identification of functional motifs, profiling genetic diseases, prediction of proteins' tertiary structure upon similarities in proteins' primary structures, etc. As a computer process, each alignment requires time and memory. In general, the time and memory requirements for performing an alignment depend of the number and length of the sequences that are aligned. In some cases, when aligning many and long sequences, such as eukaryotic chromosomes or eukaryotic complete genomes, the time and memory complexity of an algorithm may be decisive factor whether the alignment of the sequences can be performed or not on a specific computer with limited processor speed and memory. Therefore, improvements of time and memory complexity are always welcomed, but not at the cost of generating partial solutions that do not completely depict the structural, functional or evolutionary relationship between the aligned sequences.
Depending of the number of sequences that are being aligned, there are algorithms for pairwise and multiple sequence alignment. Pairwise alignment algorithms align two sequences, whereas multiple alignment algorithms align more than two sequences. In terms of the scope of the generated solution, sequence alignment algorithms can be classified as local or global. Local alignment algorithms search for the most conserved (similar) DNA fragments between two or more sequences, whereas global alignment algorithms produce solutions that incorporate each element. In terms of the conceptual framework, if we look back in history, since the pioneering work of Needleman and Wunsch [Citation1] in 1970, all algorithms for DNA pairwise alignment can be classified either as a dynamic programming based or they use some type of heuristic in order to obtain the solution (heuristic algorithms). Both groups of algorithms have their own pros and cons. Dynamic programming-based algorithms always produce a solution that maximizes the alignment score or minimizes the distance (dissimilarity) for specific metrics. This solution is referred to as an optimal one and it requires time and memory, where
and
are lengths of the sequences that are being aligned. The unfavourable quadratic time and memory complexity sometimes may limit the application of these algorithms, especially if long sequences are aligned on a standard PC configuration. In contrast to dynamic programming-based algorithms, heuristic algorithms are faster and they can be applied to long sequences, such as complete chromosomes or complete genomes. They gain on the speed of execution due to the significant reduction of the problem domain, by applying some type of filtering or prior indexing. As a result, sometimes partial or incomplete solution may be produced that does not fully represent the real structural, functional or evolutionary relationship between the sequences. A compromise between the opposite groups of algorithms is always welcomed.
The oldest, but commonly used DNA pairwise alignment algorithms, are Needleman–Wunsch [Citation1] and Smith–Waterman.[Citation2] They are dynamic-programming based implementations that maximize the score of an alignment for a specific metrics. Needleman–Wunsch [Citation1] generates global alignment, whereas Smith–Waterman [Citation2] generates local alignment in quadratic time and memory. Instead of maximizing the alignment's score, Sellers [Citation3,Citation4] minimizes the distance (dissimilarity) between two sequences. Ulam [Citation5] gave the formal definition for distance (dissimilarity) between two sequences, as the minimal number of edit operations (element deletion, element substitution or element insertion) that have to be performed in order to transform one of the sequences into the other. Smith et al. [Citation6] proved that no matter whether the alignment's score is maximized [Citation1,Citation2] or the distance (dissimilarity) between two sequences is minimized,[Citation3,Citation4] these two procedures are equivalent in terms of the structure of the generated solution under certain conditions. Rather than searching for one optimal solution, done in previous studies,[Citation1–4] algorithms of Goad and Kanehisa [Citation7] and Waterman and Eggert [Citation8] generate a list of significant local alignments. For instance, Waterman and Eggert [Citation8] identified a set of
best local alignments between two sequences. This is done by searching a dynamic programming matrix
times for
highest-scoring alignments. It is worth to mention that Fitch and Smith,[Citation9] as well as Gotoh,[Citation10,Citation11] made a distinction between the penalty that is assigned for gap opening and the penalty that is assigned for extending an already opened gap.
The first tries for linearization of the memory complexity of dynamic programming algorithms were applications of divide and conquer approach. The idea is to find in each step an interception point such as the alignment of the sequences:
and
is generated by joining the optimal alignments of the subsequences: (
and (
. This is repeated until base is aligned to base or gap. Since the path of the optimal alignment cannot pass through the north-east and south-west quadrant in dynamic programming matrix, these quadrants can be rejected in each step of the procedure. This idea was theoretically discussed by Hirschberg [Citation12] and it served as a basis for the memory linear implementations that were proposed afterwards by Mayers and Miller,[Citation13] Huang et al. [Citation14] and Huang and Miller.[Citation15]
Apart from the efforts for memory linearization, there were also attempts to improve the time complexity. The first approach is known as alignment within specific diagonal band or diagonal alignment. The idea is to calculate only those cells in the dynamic programming matrix that are located near by the main diagonal, since the path of the optimal alignment converges to it. If the length of the diagonal band equals then the time complexity can be improved up to
. This approach was discussed by Sankoff and Kruskal,[Citation16] whereas solutions of the problem were proposed by Ficket,[Citation17] Ukkonen [Citation18] and Chao et al.[Citation19] However, drawback is its application that is limited only to very similar sequences.
Heuristic algorithms were introduced to meet the necessity for significant improvement of the time complexity of DNA alignment process. They can speed up the process up to hundred times, but there is no guarantee for optimality of the generated solution. The increased speed of execution makes these algorithms suitable for comparing query sequence to database, rather than comparing only two sequences.
For instance, FASTA, the DNA and protein sequence alignment software package,[Citation20] looks for common hits between two sequences which are afterwards extended. Only 10 most significant extensions are included in the solution. Basic local alignment search tool (BLAST) [Citation21] compiles an extended list of words that resemble the words extracted from one of the sequences. These words are searched in the other sequence in order to find the perfect hits. Each hit is extended in both directions until the score drops below certain threshold. Unlike BLAST, BLAST-like alignment tool (BLAT) [Citation22] is less sensitive approach that looks for perfect or near-perfect hits, due to what can be applied only for comparison of evolutionary closely related sequences. PatternHunter [Citation23] improves the BLAST sensitivity, due to the fact that PatternHunter looks for common hits that include at least common nucleotides. Alike BLAST, these hits are afterwards extended in both directions. Fast length adjustment of short reads (FLASH) [Citation24] is also seed-based approach that looks for common words between two sequences. After calculating the differences between the starting positions of common hits, the longest list of equal differences determines the most similar fragments between the sequences. Yet Another Similarity Searcher (YASS), a pairwise sequence alignment software,[Citation25] looks for groups of close hits. The maximum distance between the hits in the groups should not exceed
where
is calculated in context to the frequencies of base substitutions and base insertions (deletions).
The most efficient approach to identify common hits between two sequences is to employ suffix tree. This data structure uses maximum unique matches-mer (MUMmer) [Citation26] and AVID, a global alignment method.[Citation27] MUMmer looks for maximum unique matches (MUMs) upon which an alignment is derived. Maximum unique match is defined as a common hit with maximum length that is not part of any other hit. By rejecting hits, whose length is less than half of the length of the longest common hit, AVID looks for a subset of non-crossing and parallel hits, which are identified by traversing suffix tree that is constructed for a sequence which is obtained by joining together the sequences being aligned. Unlike MUMmer and AVID, LAGAN, tool for large-scale multiple alignment of genomic DNA,[Citation28] detects ungapped local alignments between two sequences, where, except of nucleotide hits, mismatches are also allowed. Local ungapped alignments are identified by CHAOS algorithm, a fast database search tool that creates a list of local sequence similarities,[Citation29] which can detect short and partial hits between two sequences. LAGAN selects subset of consecutive ungapped alignments with maximum alignment score, which are afterwards connected. Super pairwise alignment (SPA) [Citation30] exploits methods based on probability and combinatorics to find the positions where gaps should be inserted. SPA calculates the percentage of local similarity within shifting window of fixed size. The shifting window includes fragments of both sequences for which the percentage of similarity is calculated as a rate between the number of matching elements and the length of the shifting window. Gaps are inserted if minimum shift of nucleotides drastically increases the percentage of local similarity.
In terms of computational aspects (time and memory complexity) and comprehensiveness of the generated solution, a compromise between dynamic programming-based algorithms and heuristic algorithms is presented. The proposed algorithm has several distinctions from the existing ones. First, the proposed algorithm does not reject any of the hits that can be included in the solution. By applying dynamic programming algorithms, some of the hits may be rejected due to the decrease of the overall alignment score for a specific metrics, in spite of the fact that these hits can be included in the alignment. The same also happens if heuristic algorithms are applied, due to the filtering in the preprocessing phase when usually shorter hits are rejected on what these algorithms gain on the speed of execution. By generating a solution that always includes the maximum possible numbers of base-to-base hits, the proposed algorithm is applicable for comprehensive structural, functional and evolutionary analysis and it can be applied for distant homology detection. Even for dissimilar sequences, the proposed algorithm is superior to dynamic programming and heuristic algorithms in terms of accurate and comprehensive detection of matching elements between two DNA samples. Second, rather than inserting gaps randomly, they are always inserted in base pairs with minimal frequency of occurrence, or in other words, we take into consideration the preservation of base pairs when determining the position where an indel is localized. Most importantly, we found that this model allows a more accurate detection of continuous indels in polyglutamine tracts of spliced mRNA of huntingtin gene than the dynamic programming. Third, unlike dynamic programming algorithms, which require
base comparisons in order to identify the set of common hits, the proposed algorithm performs less than
comparisons for the same purpose. This can be interpreted as a gain in the speed of execution, but unlike heuristic algorithms, there is no loss of data. Furthermore, if similar sequences are aligned, the time complexity of the proposed algorithm approximates to
, where
is the number of common hits (
),
is the length of the shorter sequence and
is the length of the longer sequence. When dissimilar sequences are being compared, the time performance of our algorithm equals Smith–Waterman or
time is required. Fourth, due to the memory efficient representation of each hit with data tuple that tracks the hit's starting and final positions, the memory requirement of the proposed algorithm is linear. In all tests that we performed, our algorithm required less memory than the length of the shorter sequence, regardless the identity of the sequences that were compared. This makes our algorithm one of the most memory efficient approaches proposed so far.
Materials and methods
The time and memory optimized (TMO) algorithm runs in two phases. In the first phase, we perform a search for ‘common’ and ‘consistent’ hits of two DNA sequences, upon which an alignment in the second phase is constructed.
Phase 1: searching and representing common hits
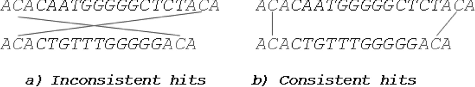
A ‘common’ hit for two DNA sequences is a character or word found in both sequences. Each hit that does not form crossing diagonal with any other hit can be considered as ‘consistent’. To introduce the concept of consistent and inconsistent common hits, the following samples are considered: and
. If the first occurrence of
in
is paired with the last occurrence of
in
, two inconsistent hits are obtained, due to the crossing diagonal that they form ((a)). Consistent hits are formed by pairing first-to-first and last-to-last occurrence of
in
and
((b)).
Figure 1. Examples of inconsistent (a) and consistent (b) common hits.
To optimize the memory requirement, a data tuple of four integers: per identified consistent hit is tracked in the memory, where
is the strating position of the hit in sequence
,
is the final position of the hit in sequence
,
is the strating position of the hit in sequence
and
is the final position of the hit in sequence
. Under this approach, tuple
represents the first consistent hit
in the samples
and
and tuples:
and
represent the second (
) and the third (
) consistent hit, respectively ().
Figure 2. Set of common and consistent hits.
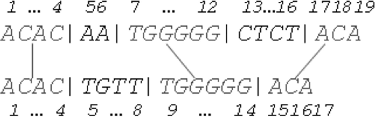
The set of consistent hits: is not the only one which can be derived for the samples that we consider. There are many other sets of consistent hits that mutually differ in the number of hits and in the number of matching nucleotides per hit, but the algorithm always tries to find the set of consistent hits which includes maximum possible number of matching elements, i.e. the set:
is reported, because there is no other set that can be defined for the samples
and
that includes more matching nucleotides than the one being detected.
During the search for consistent hits, our algorithm exploits an idea which is based on partial comparison of mismatching DNA words, instead to comparing them entirely, or nucleotides at same positions in DNA words that mismatch are mutually compared, while the first pair of mismatching nucleotides is not detected. By employing this approach, only one base comparison is required to detect the mismatch of two DNA words in the best case, whereas in the worst case, all nucleotides have to be compared.
For instance, to detect the mismatch of the words: AGCT and GCTA, only the starting nucleotides have to be compared (A to G). The remaining nucleotides are not compared, since regardless if they match or not, the words AGCT and GCTA cannot perfectly match. The worst case scenario is when all nucleotides have to be compared, in order to detect the mismatch. That is the case when all nucleotides match, except the last ones. To detect the mismatch of the words: GCTA and GCTG, four comparisons have to be performed (the number of base comparisons in this case equals the words’ length), since the last nucleotides (A and G) in these words constitute the first found pair of mismatching nucleotides. However, the number of base comparisons may be greater than one and less than the length of the words, depending of the position where the first pair of mismatching nucleotides is identified. For instance, when comparing the words TCTG and TACT, two base comparisons have to be performed in order to confirm the mismatch, since the first pair of mismatching nucleotides is found at position 2 (C to A).
Our algorithm looks for consistent hits in ‘consistent domains’. ‘Consistent domain’ is defined as a pair of sequences or subsequences in and
which are located out of identifed hits and they do not form crossing diagonal to any of the hits,
Figure 3. Searching for consistent hits: one consistent domain (a), one hit (two new consistent domains) (b), three hits (four new consistent domains) (c) and all nucleotides in consistent domains are compared (d).
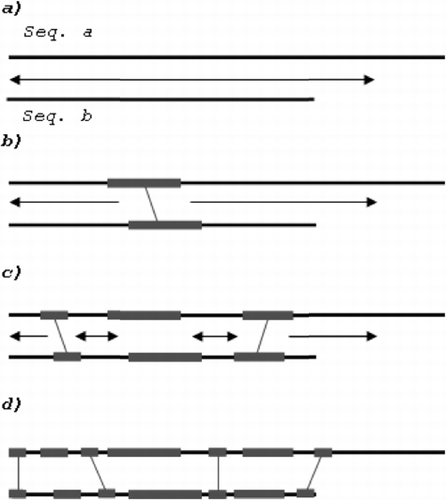
At the beginning, there is only one consistent domain which is made of the sequences being compared ((a)). The search there results in one consistent hit, which derives two new consistent domains where two new hits may be identified ((b)). These two hits together with the first identified match derive four new consistent domains, where we can also search for consistent hits ((c)) and so on until the length of at least one consistent fragment equals 0 or all nucleotides in the consistent domains are compared ((d)).
In each consistent domain, the algorithm detects the ‘longest’ and the ‘leftmost’ common hit by performing partial comparisons on mismatching DNA words of the shorter sequence/subsequence to the longer sequence/subsequence until the hit is identified.
We will discuss this approach on the short samples and
((a)). At the beginning, there is only one consistent domain which is made of the samples
and
and this is the space where the algorithm tries to find the first consistent hit. shows the order in which the algorithm compares mismatching DNA words and which nucleotides of them (highlighted) are mutually compared.
Figure 4. Identifying consistent hits for the samples and
: short samples (a), the first identified hit GCT derives two new consistent domains (b) and a single Guanine match is found in the consistent domain that follows GCT (c).
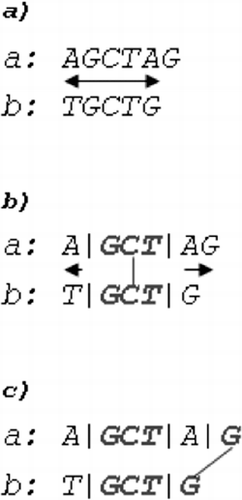
Table 1. Compared words and nucleotides, until consistent hits in the samples and
are identified.
Words of 5, 4 and 3 nucleotides from both sequences are partially compared until the first hit is identifed. The search requires totally 20 base comparisons (). The number of performed comparisons per pair of DNA words depends of the position of the first identified pair of mismatching nucleotides. For instance, one base comparison is required to detect the mismatch: (
,
since the word's starting nucleotides mismatch. Two comparisons have to be performed to detect the mismatch: (
,
, because the first mismatch of nucleotides is found at position 2 and four base comparisons are required in order to detect the mismatch of the words
and
since all nucleotides, except the last ones, match ().
The first identified hit derives two new consistent domains. The first consistent domain is made of the nucleotides that precede GCT (
in
and
in
), whereas the second is made of the nucleotides that follow GCT in
and
(
in
and
in
) ((b)). There is no common hit in the domain that precedes GCT, whereas a single guanine match is found in the consistent domain that follows GCT ((c)). The algorithm requires totally 23 comparisons to derive the set of consistent hits for the samples
and
,
} ().
Since tuples are appended into a set as hits are identified, before moving to the second phase, the set of tuples needs to be sorted according to hits’ occurrence in the analysed sequences. This means that if we have obtained the set of tuples: for the samples
and
, because
is identified at first, then
and finally
, before moving to the second phase, the algorithm will swap the first two tuples, because
corresponds to hit
that precedes
which is tracked by
. As a result, we get the set of tuples:
, which tracks the natural order of appearance of the consistent hits found in the samples
and
(
, followed by
and then
().
We can use the previous samples to analyse the gain in terms of memory reduction, compared to Huang's algorithm [Citation14] and the gain in saved nucleotide comparisons regarding Smith–Waterman.[Citation2] For instance, if Huang is applied to and
, the memory requirement would be proportional to the sum of the lengths of the sequences being aligned, i.e.
integers would have to be stored in the memory, in contrast to
integers that store the proposed algorithm for tracking hits’ positions. To derive a set of consistent hits for the samples
and
, Smith–Waterman requires
comparisons, whereas our algorithm, for the same purpose, requires 23 comparisons, without losing data for any consistent hit that may happen in heuristic algorithms for local DNA alignment, such as: FASTA,[Citation20] BLAST,[Citation21] BLAT,[Citation22] PatternHunter,[Citation23] FLASH [Citation24] and YASS.[Citation25]
Phase 2: generating local pairwise alignment
At the beginning of the second phase, the number of indels between successive hits is precisely calculated. Assuming that is the longer sequence and
is the shorter sequence, then for each two successive hits,
and
the number of gaps
which have to be inserted between them is calculated according to EquationEquation (1)
(1)
(1) , where
and
are tuples that track their positions in
and
respectively. The sign of
(>, < or 0) determines whether gaps should be inserted or not and if yes, in which mismatching fragment they have to be inserted, whether in sequence
or in sequence
.
(1)
(1)
If then
gaps have to be inserted in
. If
then
gaps have to be inserted in
. If
then no indel is found between successive hits.
If we apply EquationEquation (1)(1)
(1) to the set of consistent hits
, which was derived for the samples
and
, as a result, we obtain that two gaps have to be inserted between
and
in
and four gaps have to inserted between
and
in
, i.e.
for
and
, whereas
for
and
.
Indels/inserted gaps shift consistent hits towards their alignment. This means that in sample
needs to be shifted for two positions ((a)) and
in sample
needs to be shifted for four positions ((b)), if we want these hits to match perfectly within an alignment.
Figure 5. Indels or DNA shifts enable perfect alignment of matching fragments: TGGGGG needs to be shifted for two positions in sample a (a), ACA needs to be shifted for four positions in sample b (b).
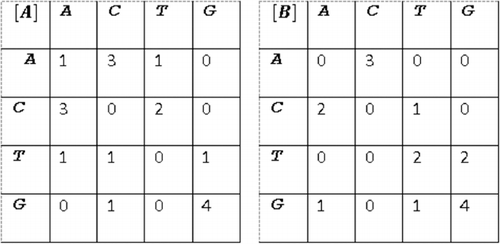
Before localizing indels, two matrices and
that track the numbers of occurrence of all nucleotide pairs
in
and
are computed. Having marked each row and each column in
and
with one of the letters:
, which correspond to the four DNA nucleotides,
represents the number of occurrence of nucleotide pair
in
. These matrices for the samples:
and
are shown in . For instance:
and
because
occurs three times in
and two times in
(). Some base pairs may not occur at all, if short sequences are analysed. Sample
conatins one
and one
nucleotide pair, but they do not occur at all in the other sample
().
Figure 6. Occurence of nucleotide pairs in
and
.
The algorithm pinpoints indels in mismatching fragments which are obtained by appending the last and the first nucleotide from surrounding hits. Extending is required, because except inside mismatching fragments, gaps can also be inserted between hits and mismatching DNA. For instance, two gaps except inside mismatch ((a)) can be also inserted between the first hit
and
in sample
((b)), as well as between
and the second hit
((c)).
Figure 7. Indels in extended mismatching fragments (marked with rectangles). Two gaps inserted: inside AA mismatch (a), between the first hit ACAC and AA (b) and between AA and the second hit TGGGGG (c).
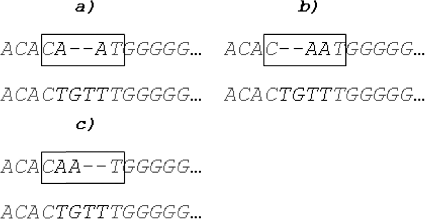
The CAAT-gapped modifications shown in are based on continuous insertion of gaps and they are three of six possible two indel transformations of the extension, since gapped transformations C–A–AT, C–AA–T and CA–A–T of CAAT, which are based on interrupted localization of indels, are also possible.
The whole process of identifying indels step by step, according to the proposed methodology, is shown in . Our algorithm reports CA–A–T modification, because AA and AT are considered for mutated triplets whose mid-elements were deleted (), due to the fact that
nucleotide pair tends to be more preserved than
/
in
(
occurs three times, whereas
and
occur only once). Therefore, rather than modifying a frequently occurring nucleotide pair (
), our algorithm will choose to pinpoint indels in rarely occurring nucleotide pairs (
/
) ((b) and (c)).
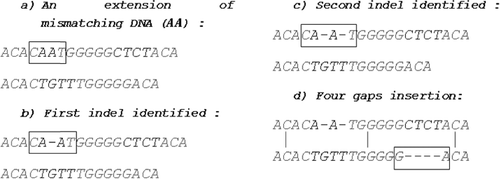
Figure 8. Steps for generating a solution: (a) CAAT is an extension of the mismatching AA fragment and contains three nucleotide pairs: CA, AA and AT; (b) gapped modification of AA; (c) gapped modification of AT; and (d) four gaps are inserted following the last nucleotide G in TGGGGG.
In general, for each extension, a list of all nucleotide pairs found there and data regarding their occurrence in is compiled. The number of occurrences of each base pair is read from matrix
(matrix
), depending on which sequence the extension belongs to. Data from the list is used to identify the rarest occurring nucleotide pair in each step, where, in fact, an indel is identified. The rarest occurring nucleotide pair
in
of all nucleotide pairs in the list is the one where the first gap is inserted
. This nucleotide pair, in which an indel is identified, has to be removed from the list before repeating the search, if another gap has to be inserted. The same is repeated until all gaps are inserted (their number is calculated according EquationEquation(1)
(1)
(1) ). If insertion has to be proceeded, but the list is empty (in all nucleotide pairs in the extension gaps were inserted), the proposed algorithm chooses to insert all remaining gaps next to the first nucleotide in the extension.
For the samples that we consider, is an extension of the mismatching AA fragment in sample
which is obtained by appending the last nucleotide C from the first hit (ACAC) and the first nucleotide T of the following hit (TGGGGG). This extension contains three nucleotide pairs:
and
((a)). The numbers of occurrence of these nucleotide pairs are read from matrix
((a)) and they are stored in list
.
Searching the list for the rarest occurring nucleotide pair in of all nucleotide pairs in the list, the algorithm pinpoints the location of the first indel. The first found nucleotide pair is always reported and that is the nucleotide pair where the gap is inserted.
The corresponding nucleotide pair in this case is in the extension
which is transformed in (CA
AT) ((b)). According to the previous discussion, the nucleotide pair
where the gap is inserted,
is removed from the list
. Once again the algorithm looks for the rarest occurring nucleotide pair in
but now to identify the position where the second gap has to be inserted. The nucleotide pair
is reported, since
appears once and CA three times. The second gap insertion transforms (CA
AT) in (CA
A
T) ((c)). Due to the absence of mismatching DNA content between
and
in
, all four gaps are inserted next to the last nucleotide
of the second hit (
) ((d)).
The algorithm pinpoints indels in ‘the least preserved nucleotide pairs’, because it assumes that they ‘originate from codons whose mid nucleotides were deleted’ when working with coding DNA. This property does not restrict the application of our algorithm only to coding DNA. It is a general approach that can also be applied to junk DNA (tandem repeats and interspersed repeats), where instead of codon mutations, mutations in non-coding triplets may be considered for accurate prediction of indels within satellite DNA or precise localization of Alu (LINE) repetitions. By applying our algorithm to eukaryotic chromosomes, we expect to retain Alu repetitions intact, due to its high occurrence rate, which can facilitate the identification of a newly evolved gene. On the other hand, Smith–Waterman will pinpoint indels in one or more Alu repetitions, if that results in a better score alignment.
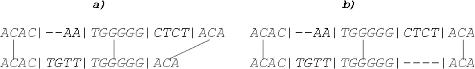
(d) shows the alignment of the samples and
according to the proposed model. As it can be noticed, all matching nucleotides are included in the solution. Shortened alignment, which partially depicts the structural relationship between the samples, is obtained if Smith–Waterman is applied, based on metrics which penalizes mismatch alignments and gap insertions with
and awards
for each nucleotide match (). For this metrics, consistent hits
and
, are not detected (included in the alignment) (). The overall alignment score would not be increased if the first consistent hit
is included in the solution. On the other hand, the overall alignment score would have been decreased for
, if the third consistent hit
is included, since four gaps have to be inserted to align three matching nucleotides:
and
.
Figure 9. Smith–Waterman solution for specific metrics.
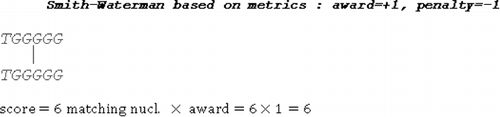
Obviously, when applying Smith–Waterman, the output depends on the selected metrics. As a consequence, some local similarities ‘remain unidentified’ (‘excluded of the alignment’). For instance, although three matching fragments () can be included in the alignment of the samples
and
, Smith–Waterman detects only one (
) matching fragment.
Partial solutions derived from Smith–Waterman may imply incorrect identification, characterization and classification of DNA data, such as errors when predicting disease predisposition of a specific gene and incorrect taxonomic classification. Unlike Smith–Waterman, the proposed algorithm maximizes the number of matching nucleotides per alignment, regardless any specific metrics and rather than inserting gaps at arbitrary positions, they are inserted at positions in the least preserved nucleotide pairs.
Due to these properties, a more accurate model for the relationship of trinucleotide repeat disorders associated with spliced mRNA can be derived in comparison to Smith–Waterman. It is known that trinucleotide repeat disorders, such as Huntington's disease,[Citation31,Citation32] Spinocerebellar ataxia [Citation33] and Dentatorubropallidoluysian atrophy [Citation34] are genetic disorders which are associated with excessive expansion of trinucleotide repeats in certain genes’ fragments. Usually, the number of uninterrupted trinucleotide repetitions determines whether an individual will be affected or not. For instance, Huntington's disease, which is associated with excessive number of uninterrupted repetitions in Huntingtin gene, causes a progressive decline of brain cells. An individual is affected if the number of uninterrupted repetitions exceeds 35. We found that indels of
repetitions in spliced mRNA of huntingtin polyglutamine (polyQ) tract are more specifically identified by the proposed algorithm than Smith–Waterman.
shows the alignments of Homo sapiens isolate hunt1 huntingtin partial coding sequence (cds) (ENA ID: EU797016.1) and Homo sapiens isolate hunt3 huntingtin partial cds (ENA ID: EU797018.1) near polyQ coding strands by applying Smith–Waterman ((a)) and the proposed algorithm ((b)). Our algorithm detects double deletion of glutamine () in polyQ tract of hunt3 huntingtin spliced mRNA regarding polyQ tract of hunt1 huntingtin spliced mRNA ((b)). On the other hand, glutamine's double deletion is not confirmed by Smith–Waterman ((a)), since non-glutamine nucleotide (cytosine) in hunt3 huntingtin partial cds is aligned to glutamine nucleotide (cytosine) in polyQ tract of hunt1 huntingtin partial cds.
Figure 10. Detection of glutamine (CAG) indels in spliced mRNA of Homo sapiens huntingtin gene by applying Smith–Waterman (a) and the proposed algorithm (b).

Regardless if non-glutamine to glutamine or non-glutamine to non-glutamine cytosine is aligned, the overall Smith–Waterman alignment score is not affected and that is the only parameter, which Smith–Waterman considers. The proposed algorithm does not separate cytosine in hunt3 huntingtin partial cds from …CCTCAAGTCCTT matching fragment, because its length is increased for one, if non-glutamine cytosines are aligned ((b)). Moreover, the nucleotide pair is considered to be unsplittable due to its frequent occurrence in hunt3 huntingtin partial cds ((b)).
By knowing the number of uninterrupted glutamine repetitions in hunt1 huntingtin spliced mRNA, it is easy to find out whether the carrier of spliced mRNA, associated with hunt3 huntingtin, is affected by the disease or not.
In addition, we summarize the instructions in which our algorithm runs. Following these instructions, we developed a desktop application in C# which was tested on Acer Aspire 5570Z computer with Genuine Intel CPU 2080 at 1.73 GHz and 2 GB RAM memory.
Input: DNA sequence , DNA sequence
Output: Local alignment A
10. Partly comparing mismatching DNA words, IDENTIFY ALL CONSISTENT HITS.
20. REPRESENT each hit with data tuple: in the memory.
30. SORT the set of tuples according to their appearance in and
.
40. CALCULATE the number of indels between successive hits by applying EquationEquation (1)(1)
(1) .
50. TRACK the number of occurrences of each nucleotide pair in
in matrix
(
).
60. EXTEND each mismatching fragment (where at least one indel is identified) with the last nucleotide from the preceding hit and the first nucleotide from the following hit.
70. FOR EACH nucleotide pair in extension, read the number of occurrence from matrix (
) and store it in list
.
80. UNTIL all gaps are inserted.
90. SEARCH for the rarest occuring nucleotide pair
and TRANSFORM
in
in the extension.
100. PRINT alignment A.
is a screen capture of the application output, when we compared partial cds of homosapiens hunt1 huntingtin (ENA ID: EU797016.1) with hunt3 huntingtin (ENA ID: EU797018.1) near polyQ tracts, where indels / gaps are marked with ‘N’.
Figure 11. Output of the application near polyQ tracts of Homo sapiens huntingtin hunt1 to Homo sapiens huntingtin hunt3 partial cds alignment.

In total, there were nine tests that were performed on very similar huntingtin partial cds (ENA IDs: EU797016–24.1), which were retrieved from the European Nucleotide Archive,[Citation35] as well as a test, which was performed on dissimilar coding DNA of human tetratricopeptide repeat protein 25 (UniProt ID: Q96NG3) to tetratricopeptide repeat protein 23 (UniProt ID: Q5W5×9). The same samples were also aligned using European Molecular Biology Open Software Suite (EMBOSS) Water implementation of Smith–Waterman.[Citation36]
Both algorithms were compared in terms of computational performance and hit detection rate. Additionally, we analyzed the number of detected glutamine deletions in polyglutamine tracts and the number of detected matching residues in tetratricopeptide repeats (TPR), which are considered to be similar, but different. TPR is usually made of 34 amino acids and in spite of the fact that consensus has been derived, all residues, except alanine at positions 20 and 27 (
), are likely to change.
Results and discussion
We got better computational results when we analysed partial cds of huntingtin gene than when we analysed the coding DNA of rather than similar tetratricopeptide repeat proteins. Even for tetratricopeptide repeat proteins, for which the percentage of identity is approximately 24%, our algorithm requires less memory than Huang, but the time complexity seems to approximate Smith–Waterman, i.e. in both cases, six seconds were required for deriving a solution. In all cases, regardless if similar or dissimilar sequences were analysed, our algorithm detected more consistent hits than Smith–Waterman that has resulted in better detection of matching residues in tetratricopeptide repeats. Moreover, our algorithm pinpointed indels in polyglutamine tracts of partial cds of huntingtin gene more accurately than Smith–Waterman's due to the fact that it considers the relative frequency of mismatching DNA, when it pinpoints an indel.
We chose to compare the performance of our algorithm to Smith–Waterman and Huang, because Smith–Waterman is considered to be the most accurate method for local pairwise alignment, whereas Huang has the same accuracy of Smith–Waterman, but instead of memory, it requires linear
space for deriving a solution. There are computationally better algorithms than Smith-Waterman/Huang, such as: FLASH, YASS and PatternHunter, but in most of the cases they generate partial (incomplete) solutions.
Glutamine deletions in polyQ tracts were analysed by applying the proposed algorithm and Smith–Waterman based on and
metrics. We found that a total of 17 missing glutamines in polyQ tracts were more specifically identified by the proposed algorithm compared to Smith–Waterman (). The number of precisely identified deletions in polyQ tracts applying the proposed algorithm ranges between 0 and 3 per alignment . Due to the key algorithmic feature to preserve highly conserved
and
nucleotide pairs in uninterrupted
repetitions in polyQ tracts, in 9 out of 10 cases our algorithm detects at least one more glutamine deletion more specifically than Smith–Waterman. Equal algorithmic outputs in the same context are obtained only once ().
Table 2. Detected glutamine deletions in polyQ tract by applying the proposed algorithm.
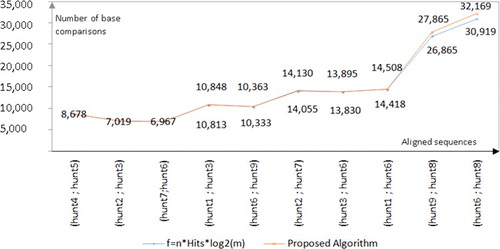
In the cases of spliced hungtingtin mRNA alignments, our algorithm runs faster than Smith–Waterman, due to the fewer numbers of comparisons that were performed when compared with Smith–Waterman. The number of performed comparisons and the overall time required per alignment, when applying our algorithm and Smith–Waterman, are given in . Less nucleotide comparisons are performed, because rather than comparing mismatching DNA entirely, our algorithm escapes performing redundant comparisons, starting from the first found pair of mismatching nucleotides. According to the data in , the speed up of the proposed algorithm over Smith–Waterman depends on the number of matching fragments (hits). The speed up is greater, if fewer, but longer hits are detected (). In the best case, our algorithm runs 23 to 27 times faster than Smith–Waterman. These numbers correspond to alignments which include only two matching fragments. In the worst case, the seed up is approximately five times faster and that is the case when 8(9) hits are detected (). In spite of the fact that most of the time is spent for comparing nucleotides, minor part of the total time is spent for computing the frequencies of mismatching DNA and tracking/sorting the set of consistent hits. For hunt 4 (EU797019.1) and hunt 5 (EU797020.1) partial cds, it took only 0.1%, while 99.9% of the total time was required for comparing nucleotides.
Table 3. Time performance analysis.
On the other hand, six seconds were required for computing the alignment of human tetratricopeptide repeat protein 25 (UniProt ID: Q96NG3) to human tetratricopeptide repeat protein 23 (UniProt ID: Q5W5×9), when applying our algorithm and Smith–Waterman. In this case, there was no speed up over Smith–Waterman, because instead of few, but long common hits, our algorithm detected 162 short identities, which were not longer than four residues. The presence of many, but short common hits, as in this case, has a negative impact on the overall time performance, mainly due to the sizeable number of nucleotide comparisons that had to be performed. In this case, we estimated that 0.8% of the total time was spent for computing the frequencies of mismatching DNA and tracking/sorting the set of consistent hits that was likely to happen due to the high portion of mismatching DNA and the increased length of the consistent hits set.
We got better results than Smith–Waterman for the pairwise alignments of partial cds of huntingtin gene, because there were only few, but long common hits (2–9). This is specific only for very similar DNA data (the percentage of identity for partial cds of huntingtin gene is more than 90%), for which the obtained results confirm that our algorithm runs several times faster than Smith–Waterman. We found that is the best fitting function for the time samples of spliced huntingtin mRNA alignments and, therefore, the time complexity of our algorithm when it works with very similar DNA is
, where
is the length of the longer sequence,
is the length of the shorter sequence and
is the number of hits (matching fragments) in alignment ().
Figure 12. Best fitting function for the number of nucleotide comparisons.
On the other hand, if sequences with poor identity are analysed, such as the cds of human tetratricopeptide repeat protein 23 to human tetratricopeptide repeat protein 25, for which the percentage of identity is 24%, our algorithm is not worse than Smith–Waterman, but there is no time improvement ( time is required).
The results of the memory analysis for partial huntingtin cds are shown in . Space requirements of the proposed algorithm and Huangs’ memory linear algorithm are compared. The memory requirement of the proposed algorithm was calculated according to EquationEquation (2)(2)
(2) and it depends on the
and the size of the list
which tracks the numbers of occurrence of all nucleotide pairs in mismatching extensions. The constant
in EquationEquation (2)
(2)
(2) represents the cost for storing two matrices:
and
in the main memory.
(2)
(2)
Table 4. Memory analysis.
According to the results in , our algorithm requires between 8 and 20 times less memory than Huang. The same relationship, in terms of the number of hits, can be derived again, i.e. fewer hits are detected and the memory saving is greater (). The alignment which includes nine hits is the worst case of memory improvement, whereas the best one corresponds to the alignment of only two hits
The proposed TMO algorithm required 2,592 B (648 integers) for tracking 162 residual common hits of human tetratricopeptide repeat protein 23 to human tetratricopeptide repeat protein 25. There were 1 four-residue hit, 3 three-residue hits, 25 two-residue hits and 133 one-residue hits detected. Regardless of the common hit length, our algorithm requires 16 B (four integers) per match representation, which means that the cost for tracking four-residues common hit equals the cost for tracking one-residue common hit or in both cases, 16 B (four integers) are required for tracking the hits' locations. For the same purpose, Huang required five times more memory, or 13,428 B. This confirms the space efficiency of the proposed methodology, even when sequences with poor percentage of identity are analysed.
Further improvement of the memory complexity for for tracking data tuples is possible, if instead of four integers per data tuple, we use data tuples of three integers, since each common hit can be tracked with only three integers
, where
is the starting position of the common hit in sequence
,
is the starting position of the common hit in the sequence
and
is the length of the common hit, which is the same in both sequences. This is the maximum possible cut per data tuple without losing any tracking information, but compared to the first representation, which is based on four integers
, this results in an additional memory saving of 25% for tracking data tuples.
If we exclude the length of the hits from the data tuples, the number of indels between successive hits can also be precisely calculated, but this will result in memory saving of 50% for tracking tuples, compared to the first representation. This requires rearrangement of EquationEquation (1)
(1)
(1) , such as the final positions of the
hit in
,
have to be written as
(
, where
is the starting position of the
hit in
and
is the length of the hit. With a substitution of
with
(
in EquationEquation (1)
(1)
(1) , we get EquationEquation (3)
(3)
(3) , or the starting positions of hit k and hit k+1 in
and
and
are enough to calculate the number of indels.
(3)
(3)
By using EquationEquation (3)(3)
(3) , instead of EquationEquation (1)
(1)
(1) , we reduced the overall memory requirement with up to 20% for huntingtin spliced mRNA () and with 35% in the case of human tetratricopeptide repeat protein 25 to tetratricopeptide repeat protein 23 alignment. We got better memory reduction ratio for human tetratricopeptide repeat proteins, because there were more hits that were identified (162) than in the cases of huntingtin spliced mRNA alignments (2–9). The overall memory complexity was not reduced for 50%, because according to EquationEquation (2)
(2)
(2) , the algorithm also needs memory for tracking the mismatching DNA (|L|) and 32 integers for two matrices
and
.
An equal number of matching nucleotides in spliced mRNA samples of huntingtin gene is identified when applying the proposed algorithm and EMBOSS Water implementation of Smith–Waterman based on and
metrics. But when we run tests based on increased gap penalty (
,
), some of the matching nucleotides which were detected by the proposed algorithm were not detected by applying Smith–Waterman (). Regardless of any specific alignment metrics, in 9 out of 10 cases, our algorithm detected more matching nucleotides in partial cds of huntingtin gene than Smith–Waterman (). Their number ranges between 8 and 19 and they are excluded by Smith–Waterman, due to the decrease the overall alignment score.
Table 5. Number of matching nucleotides per alignment.
When we compared human tetratricopeptide repeat protein 23 to human tetratricopeptide repeat protein 25, 59 matching amino acids were found by Smith–Waterman, whereas our algorithm reported 192. The analysis of the number of matching residues in tetratricopeptide repeats: TPR1, TPR2, TPR3 and TPR4 of human tetratricopeptide repeat protein 23 to human tetratricopeptide repeat protein 25 shows that our algorithm detected 46 matching amino acids more in tetratricopeptide repeats than Smith–Waterman ().
Table 6. Detailed analysis of matching residues in TPR1–TPR4 repeats.
Moreover, Smith–Waterman completely excluded TPR4 repeat from the solution, due to the fact that it could not detect even a single matching residue, whereas our algorithm detected 14 matching amino acids there (). Our algorithm detected between 11 and 12 matching residues more in TPR1, TPR2 and TPR3 than Smith–Waterman and a total of four consensus elements were confirmed.
Even for sequences with small percentage of identity, such as human tetratricopeptide repeat protein 23(25), our algorithm was able to detect more common hits than Smith–Waterman, regardless of any specific alignment metrics what makes it suitable for comprehensive analysis of structural, functional and evolutionary relationship between coding DNA data.
The problem of identifying common hits between two DNA sequences has been widely explored in the science and different solutions have been proposed. Index-based solutions, such as MUMmer, AVID and FLASH, can discover common hits faster than our algorithm, but they are expected to be worse in terms of memory complexity, due to the indexed data structure that they employ. For instance, MUMmer and AVID build suffix tree, whose size is proportional to the sum of the sequences’ lengths and FLASH builds an index of all words found in both sequences, regardless if they are matching or not. There is also a preprocessing phase which precedes the actual hits' searching phase, which may be considered an additional computational cost.
Recent methods based on DNA hashing enable fast comparison on DNA words and efficient data storage. Instead of comparing words, their hash representations are mutually compared and the cost for storing hash of a DNA word is only 4 B (1 integer). Sequence Search and Alignment by Hashing Algorithm (SSAHA) [Citation37] is one of the hash-based implementations which can be applied for discovering common hits based on their hash-representations. It assigns a unique number to each DNA nucleotide 0, 1, 2 or 3},
and calculates the hash of DNA word of
nucleotides
by applying the equation
. SSAHA requires 4 B (1 integer) for storing DNA hash
, 4 B (1 integer) for hash's starting position
and 4 B (1 integer) for the index
of the sequence where the hash occurs, or 12 B (3 integers) per hashed/tracked DNA word are required. This means that for hashing/tracking 12 Kbp common hit
B (7,200 integers) would be required, given that
. For tracking the same hit, our algorithm requires 16 B (four integers), which results in memory saving of
B.
In general, indexing and hashing allow faster comparison of DNA words, but there is an additional memory cost for storing word's index/hash. For instance, it would be faster to compare hashes GCTAA
and
GCTAG) than performing partial comparison based on linear walk on GCTAA and GCTAG, because instead of comparing five characters (5 B), integer would have to be compared with integer, for what is a required comparison of 4 B. Faster comparison, based on indexing/hashing, is only possible at the cost of increased memory requirement, because 8 B (two integers) that track the hashes of GCTAA and GCTAG would have to be stored in the memory. There is also an additional computational cost for computing the hashes of the words, which is proportional to the length of words that are indexed. Our algorithm is a compromise between the time and memory requirement, i.e. it is expected to run slower than hash/index-based methods, but there is no cost for storing DNA index/hash and no preprocessing phase that precedes the actual hits' searching phase.
Theoretically, different sets of consistent hits can be defined if at least one of the common hits is found as a repeating sequence. For instance, if the trinucleotide GCT in the sample is extended in
trinucleotide microsatellite (
, then, instead of one, three different sets of consistent hits, which are equal in terms of the matching content:
},
} and
} can be defined for the samples
and
. So, which one of the three candidates does the algorithm report?
Our algorithm works accurately for coding and non-coding DNA, due to the fact that the leftmost positioned consistent hits in minisatellite (microsatellite) DNA are always reported. For the samples and
the first set:
} is the one which is reported, because
is the leftmost positioned microsatellite triplet of all three (
,
and
in the sample
((a)). This approach enables accurate indel's prediction, since the alignment is built starting from the leftmost positioned match in the set of consistent hits. Our model is based on the set:
} where two
indels are pinpointed ((a)). Incorrect solutions would have been derived if the second (third) candidate set of consistent hits were used, where instead of 2, 1(0)
local indels are predicted ((b) and 13(c)). summarizes the key features of our algorithm.
Figure 13. Aligning sequences with repeats. Hits in the microsatellite DNA: GCT (2,4) – being reported (a), GCT (5,7) (b) and GCT (8,10) (c).
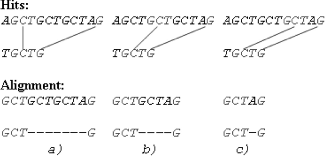
Table 7. Summary of the proposed algorithm's key features.
Conclusions
In the present study, a TMO and more accurate algorithm than the well-known and often used dynamic programming algorithms is presented. Four crucial aspects outline the benefit of the proposed algorithm. First, our algorithm allows precise detection of missing glutamine-coding triplets in polyglutamine tracts of partial cds of huntingtin gene, due to its predisposition to pinpoint indels in less frequently occurring nucleotide pairs. This property makes our algorithm suitable for accurate detection of Huntington's disease and other diseases, which are associated with excessive trinucleotide repetitions, such as: Spinocerebellar ataxia and Dentatorubropallidoluysian atrophy. Second, our algorithm runs faster than Smith–Waterman for very similar DNA, but not at the cost of generating partial or shortened alignments, which may happen in recent developments, such as FLASH, YASS, PatternHunter and other related approaches that usually cut in the search space in order to improve the time performance. It is also very important to note that the time improvement (the reduced number of comparisons) does not affect the completeness of the solution. Third, our algorithm requires less memory than the most memory efficient dynamic programming algorithm, the Huang's algorithm, regardless if similar or dissimilar DNA is analysed. The memory complexity of the proposed algorithm is linear and it is proportional to the number of hits being detected. Fourth, unlike Smith–Waterman, our algorithm does not reject any of the matching nucleotides which can be included in the solution and, therefore, is suitable for a complete detection of the homology between two DNA samples.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48(3):443–453.
- Smith T, Waterman M. Identification of common molecular subsequences. J Mol Biol. 1981;147(1):195–197.
- Sellers PH. An algorithm for the distance between two finite sequences. J Combin Theory Ser A. 1974;16(2):253–258.
- Sellers PH. The theory and computation of evolutionary distances: pattern recognition. J Algorithms. 1980;1(4):359–373.
- Ulam SM. Some combinatorial problems studied experimentally on computing machines. In: Zaremba SK, editor. Applications of number theory to numerical analysis. San Diego, CA, Academic Press; 1972. p. 1–3.
- Smith TF, Waterman MS, Fitch WM. Comparative biosequence metrics. J Mol Evol. 1981;18(1):38–46.
- Goad WB, Kanehisa MI. Pattern recognition in nucleic acid sequences. I. A general method for finding local homologies and symmetries. Nucleic Acids Res. 1982;10(1):247–263.
- Waterman MS, Eggert M. A new algorithm for best subsequence alignments with application to tRNA-rRNA comparisons. J Mol Biol. 1987;197(4):723–728.
- Fitch WM, Smith TF. Optimal sequence alignments. Proc Natl Acad Sci U S A. 1983;80(5):1382–1386.
- Gotoh O. An improved algorithm for matching biological sequences. J Mol Biol. 1982;162(3):705–708.
- Gotoh O. Pattern matching of biological sequences with limited storage. CABIOS. 1987;3(1):17–20.
- Hirschberg DS. A linear space algorithm for computing maximal common subsequences. Commun ACM. 1975;18(6):341–343.
- Myers EW, Miller W. Optimal alignments in linear space. CABIOS. 1988;4(1):11–17.
- Huang X, Hardison RC, Miller W. A space-efficient algorithm for local similarities. CABIOS. 1990;6(4):373–381.
- Huang X, Miller W. A time-efficient, linear-space local similarity algorithm. Adv Appl Math. 1991;12(3):337–357.
- Sankoff D, Kruskal JB. Time warps, string edits, and macromolecules: the theory and practice of sequence comparison. 1st ed. Boston, MA: Addison-Wesley; 1983. p. 382.
- Fickett JW. Fast optimal alignment. Nucleic Acids Res. 1984;12(1):175–179.
- Ukkonen E. Algorithms for approximate string matching. Inf Control. 1985;64(1):100–118.
- Chao KM, Pearson WR, Miller W. Aligning two sequences within a specified diagonal band. CABIOS. 1992;8(5):481–487.
- Lipman DJ, Pearson WR. Rapid and sensitive protein similarity searches. Science. 1985;227(4693):1435– 1441.
- Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410.
- Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12(4):656–664.
- Ma B, Tromp J, Li M. PatternHunter: faster and more sensitive homology search. Bioinformatics. 2002;18(3):440–445.
- Califano A, Rigoutsos I. FLASH: A fast look-up algorithm for string homology. In: IEEE, editor. Proceedings of IEEE Conference on Computer Vision and Pattern Recognition; 1993 Jun 15–17; New York, NY: IEEE Computer Society; 1993.
- Noé L, Kucherov G. YASS: enhancing the sensitivity of DNA similarity search. Nucleic Acids Res. 2005;33(suppl 2):W540–W543.
- Delcher AL, Kasif S, Fleischmann RD, et al. Alignment of whole genomes. Nucleic Acids Res. 1999;27(11):2369–2376.
- Bray N, Dubchak I, Pachter L. AVID: A global alignment program. Genome Res. 2003;13(1):97–102.
- Brudno M, Do CB, Cooper GM, et al. NISC comparative sequencing program. LAGAN and multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003;13(4):721–731.
- Brudno M, Morgenstern B. Fast and sensitive alignment of large genomic sequences. In: IEEE, editor. Bioinformatics Conference; 2002 Aug 14–16; Stanford, CA: IEEE Computer Society; 2002.
- Shen SY, Yang J, Yao A, et al. Super pairwise alignment (SPA): an efficient approach to global alignment for homologous sequences. J Comput Biol. 2002;9(3):477–486.
- Walker FO. Huntington's disease. The Lancet. 2007;369(9557):218–228.
- Craufurd D, Thompson JC, Snowden JS. Behavioral changes in Huntington disease. Cogn Behav Neurol. 2001;14(4):219–226.
- Orr HT, Chung MY, Banfi S, et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4(3):221–226.
- Nagafuchi S, Yanagisawa H, Sato K, et al. Dentatorubral and pallidoluysian atrophy expansion of an unstable CAG trinucleotide on chromosome 12p. Nat Genet. 1994;6(1):14–18.
- The European Nucleotide Archive [Internet]. Heidelberg: The European Molecular Biology; [cited 2015 June 20]. Available from: http://www.ebi.ac.uk/ena/.
- EMBOSS Water online tool [Internet]. Heidelberg: The European Molecular Biology; [cited 2015 June 20]. Available from: http://www.ebi.ac.uk/Tools/psa/emboss_water/.
- Ning Z, Cox AJ, Mullikin JC. SSAHA: a fast search method for large DNA databases. Genome Res. 2001;11(10):1725–1729.