
ABSTRACT
Isodon rubescens is an important medicinal plant in China that has been shown to reduce tumour growth due to the presence of the compound oridonin. In an effort to facilitate molecular research on oridonin biosynthesis, we reported the use of next generation massively parallel sequencing technologies and de novo transcriptome assembly to gain a comprehensive overview of I. rubescens transcriptome. In our study, a total of 50,934,276 clean reads, 101,640 transcripts and 44,626 unigenes were generated through de novo transcriptome assembly. A number of unigenes – 23,987, 10,263, 7359, 18,245, 17,683, 19,485, 9361 – were annotated in the National Center for Biotechnology Information (NCBI) non-redundant protein (Nr), NCBI nucleotide sequences (Nt), Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO), Swiss-Prot, protein family (Pfam), gene ontology (GO), eukaryotic ortholog groups (KOG) databases, respectively. Furthermore, the annotated unigenes were functionally classified according to the GO, KOG and KEGG. Based on these results, candidate genes encoding enzymes involved in terpenoids backbone biosynthesis were detected. Our data provided the most comprehensive sequence resource available for the study on I. rubescens, as well as demonstrated the effective use of Illumina sequencing and de novo transcriptome assembly on a species lacking genomic information.
Introduction
Isodon rubescens is an important perennial plant and medicinal herb belonging to the Isodon genus and Lamiaceae family. It is distributed in Henan, Shanxi, Hunan and Guizhou provinces, where it is usually referred to by the common name donglingcao. There has been a documented medicinal use of the plant in the Taihang Mountains in Henan province since 1972. It has been shown to possess antimicrobial and antitumour activities.[Citation1] These properties have resulted in the widespread study of the plant by scholars in China and abroad.
More than 20 terpenoids, a variety of triterpenoids, monoterpenes and phenolic acids have been isolated from I. rubescens.[Citation2–8] Many studies have shown oridonin to be the main compound that possesses remarkable anticancer, antiinflammatory and antibacterial properties.[Citation9–11] The recognized medicinal value of I. rubescens is driving researches of its tissues, cells culture and biology.[Citation12–16] While there have been numerous reports on the chemical composition, pharmacological effects and clinical efficacy of I. rubescens, genomic data and information are scarce.[Citation17] In order to gain insights into terpenoid biosynthesis, especially that of diterpenoid, characterization of functional genes via genome sequencing of the transcriptome is necessary. RNA sequencing (RNA-seq) has been widely used for de novo transcriptome sequencing to detect gene expression and other useful genomic information in many plant species without prior genomic information.[Citation18–23] Despite its obvious potential, next generation sequencing methods have not yet been applied to I. rubescens research.
In this study, RNA-seq was used to analyse the transcriptome of I. rubescens by using Illumina paired-end sequencing technology on a HiSeq 2000 platform to discover candidate genes that encode enzymes involved in terpenoid biosynthesis.
Materials and methods
Plant material and RNA extraction
I. rubescens’ leaves were collected from plants growing at standardized planting base in Jiyuan City, Henan province in October 2014. Total RNA was extracted using a TRIzol Kit (Promega, USA). The purity and concentration of the isolated RNA were checked using a Spectrophotometer (NanoPhotometer®, Implen, CA, USA) and an RNA Assay Kit (Qubit® 2.0 Flurometer, Life Technologies, CA, USA).
Transcriptome sequencing and de novo assembly
A total of 3 μg RNA per sample were used as input material for the RNA sample preparations. Sequencing libraries were generated using NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer's recommendations, and index codes were added to attribute sequences to each sample. Briefly, mRNA was purified from total RNA by using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5X). First-strand cDNA was synthesized using a random hexamer primer and M-MuLV Reverse Transcriptase (RNaseH-). Second-strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. The remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of the 3′ ends of DNA fragments, a NEBNext Adaptor with a hairpin loop structure was ligated to prepare for hybridization. In order to select cDNA fragments of preferentially 150–200 bp in length, the library fragments were purified with AMPure XP system (Beckman Coulter, Beverly, USA). Then, 3 μL USER Enzyme (NEB, USA) was used with size selected, adaptor-ligated cDNA at 37 °C for 15 min, followed by 5 min at 95 °C before polymerase chain reaction (PCR). PCR (ABI9700, USA) was performed under the following conditions: 98 °C for 30 s, 98 °C for 10 s, 65 °C for 10 s (16 cycles), followed by 72 °C for 30 s and 72 °C for 5 min. The quantitative PCR reactions (50 μL) consisted of 23 μL DNA fragment linking adaptor, 1 μL univesial primer, 1 μL NEBNext® index (X) primer for Illumina and 25 μL High-Fidelity 2 X PCR Master Mix. Finally, the PCR products were purified (AMPure XP system) and the library quality was assessed on the Agilent Bioanalyzer 2100 system. A paired-end Illumina sequencing was performed on a HiSeq 2000 platform. The Illumina reads were then assembled into contigs and unigenes using Trinity software (version: v2012-10-05).[Citation24]
Gene functional annotation
Gene function was annotated based on the following databases: National Center for Biotechnology Information (NCBI) non-redundant protein (Nr), NCBI nucleotide sequences (Nt), protein family (Pfam), Swiss-Prot (a manually annotated and reviewed protein sequence database), eukaryotic ortholog groups (KOG), Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO), gene ontology (GO), by using the nucleotide basic local alignment search tool (BLAST) software.
Coding DNA sequences (CDS) prediction
If a unigene had no alignment hit (match) in the above databases, CDSs were predicted by BLASTX and Expressed Sequence Tag (EST) Scan software (3.0.3).
Quantitative reverse-transcription PCR (qRT-PCR) analysis
1-Deoxy-D-xylulose-5-phosphate reductoisomerase (DXR), 1-deoxy-D-xylulose-5-phosphate synthase (DXS), ent-kaurene synthase (KS), ent-kaurene oxidase (KO), ent-kaurenoic acid hydroxylase (KAO), related to terpenoid backbone and diterpenoid biosynthesis, were chosen to determine the expression patterns in root, stem and leaf samples of I. rubescens. qRT-PCR was performed on an ECO™ real-time PCR system (Illumina, USA) with the SsoFastTM EvaGreen supermix (Bio-Rad, USA) under the following conditions: 95 °C for 10 min, followed by 40 cycles of 95 °C for 10 s, 55 °C for 30 s and 72 °C for 15 s. For the quantitative RT-PCR, the reaction (20 μL) consisted of 2 μL first-strand cDNA, 10 μL QuantiNova SYBR Green PCR Master Mix (2×) (QIAGEN), 0.4 μL of each of the used primers and 7.6 μL ddH2O. The expression levels of different genes were normalized to the constitutive expression level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primers and their sequences were as follows: DXR – forward (CTTGTGTGCAAGAGGGAGAA), DXR – reverse (TCACTGGAATTGTTGGCTGT), DXS – forward (CATGGTCTACGGGTGACAGT), DXS – reverse (TCTTCGACAGTGATCAAGGC), KS – forward (ACATGGGTGCCAACTATTCA), KS – reverse (GGGTGAACAAACCGGTCTAC), KO – forward (TGCAATCAGTTTCATGCTCA), KO – reverse (GACGCCTAGTTCCTCCACAT), KAO – forward (CCTAGGCAGTTATCCTTCGG),KAO – reverse (GATTGTGCCCTGGAAATGAT), GAPDH – forward (AAACGCCTAACTTCGCATCT), GAPDH – reverse (CCCGAGTATTGGACCTGATT).
Statistical analyses
Each treatment was performed in three replicates. Analysis of variance (ANOVA) was used for data evaluation.
Results and discussion
Transcriptome assembly
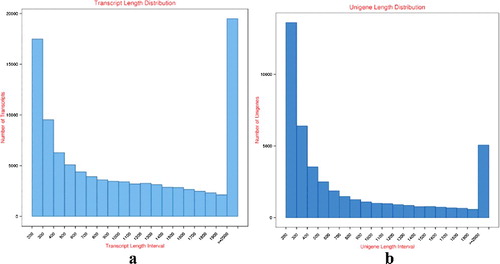
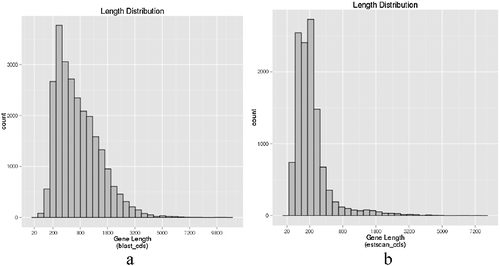
After performing the paired-end Illumina sequencing, 52,921,428 raw reads were obtained. By removing the adapter containing reads, ploy-N containing reads and low quality reads from the raw data, 50,934,276 clean reads were generated. The Q20 percentage (proportion of nucleotides with quality value larger than 20 in reads; sequencing error rate < 1%) and Guanine and Cytosine nucleotides (GC) percentage (proportion of guanidine and cytosine nucleotides among the total nucleotides) were 98.49% and 48.06%, respectively (). Based on clean reads, Trinity software was adopted to piece together all the reads. As a result, 101,640 transcripts (from 201 to 10,478 bp) with an average of 1227 bp were obtained ((a)). In addition, a total of 44,626 unigenes (mean length of 876 bp), with a total of 39,092,024 nucleotides were yielded and 12,922 unigenes (28.96%) were longer than 1000 bp ((b)).
Table 1. Summary of sequencing output statistics.
Figure 1. Length distribution of transcripts (a) and unigenes (b) of I. rubescens.
Functional annotation of the unigenes
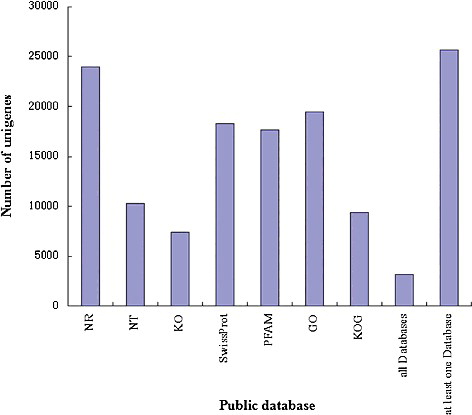
To obtain complete annotations, the gene functions were annotated based on the following databases: NCBI Nr, Nt, Pfam, Swiss-Prot, KOG, KEGG Orthology (KO) and GO. It can be seen from that a total of 23,987 (53.75%) unigenes were annotated in Nr, 10,263 (22.99%) unigenes were annotated in Nt, 7359 (16.49%) unigenes were annotated in KO, 18,245 (40.88%) unigenes were annotated in Swiss-Prot, 17,683 (39.62%) unigenes were annotated in Pfam, 19,485 (43.66%) unigenes were annotated in GO, 9361 (20.97%) unigenes were annotated in KOG, 3198 (7.16%) unigenes and 25,678 (57.54%) unigenes were annotated in all databases and in at least one database, respectively. These results showed that the Illumina-based sequencing project yielded an extensive proportion of the diverse genes expressed in I. rubescens. We also found that 42.46% of the unigenes were not annotated in all databases, meaning that novel genes probably will be found in I. rubescens.
Figure 2. Database distribution of unigenes’ functional annotations.
Gene ontology classification
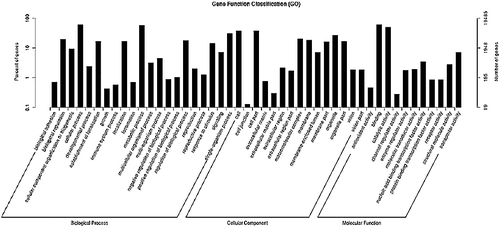
GO assignments were used to classify the functions of the predicted I. Rubescens genes. Based on sequence homology, 19,485 unigenes were categorized into three functional groups, including three ontology categories: biological process, molecular function and cellular component. shows that a high percentage of genes fall under the biological process category and are involved in cellular processes (11,799) and metabolic processes (11,236). This finding provides abundant information on novel genes involved in the metabolic pathways, including secondary metabolism. For the cellular component category, cell (7329) and cell part (7314) were prominently represented, followed by organelle (5318), macromolecular complex (3909), membrane (3620) and organelle part (3160). Within the molecular function category, binding (11,879) and catalytic activity (9931) represented the majority of unique sequences (). In conclusion, these GO annotations provided comprehensive information on specific biological processes, molecular functions and cellular structures of I. Rubescens’ transcripts. These results may lead to the identification of novel genes involved in secondary metabolite synthesis pathways.
Figure 3. Distribution of I. rubescens unigenes among the GO functional classes.
Functional classification by KOG
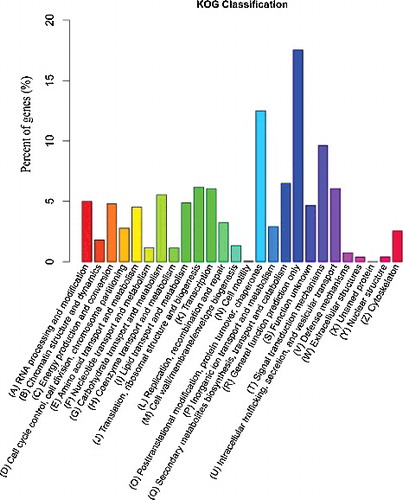
In order to evaluate further the function of the assembled unigenes, we searched the annotated unigenes involved in KOGs. Out of 23,987 Nr hits, 9361 unigenes were classified into 26 groups (). Among the 26 KOG categories, the largest classification was general function (1642 or 17.54%), followed by post-translational modification (1170 or 12.50%), signal transduction mechanisms (902 or 9.64%) and secondary metabolite biosynthesis, transport and catabolism (609 or 6.51%). The most abundant sequences in this category were for cytochrome P450 (COG2124), with a total of 84 unigenes involved in various metabolic pathways. This was followed by intracellular trafficking, secretion and vesicular transport (568 or 6.06%), transcription (567 or 5.89%), carbohydrate transport and metabolism (518 or 5.53%) and energy production and conversion (448 or 4.78%). Other categories were replication, recombination and repair (305 or 3.26%), inorganic ion transport and metabolism (273 or 2.92%), cell cycle control, cell division, chromosome partitioning (261 or 2.79%), chromatin structure and dynamics (169 or 1.81%), cell wall/membrane/envelope biogenesis (125 or 1.34%), nucleotide transport and metabolism (110 or 1.18%), coenzyme transport and metabolism (108 or 1.18%) and nuclear structure (40 or 0.43%). The following categories represented the smallest groups: defense mechanisms (71 or 0.76%), extracellular structures (38 or 0.405%) and nuclear structure (40 or 0.43%). It is worth noting that 437 or 4.67% and 1 or 0.01% unigenes were annotated about function unknown and unnamed protein, respectively, which indicated that new genes were likely involved in the secondary metabolites’ biosynthesis.
Figure 4. Functional classifiation of I. rubescens unigenes according to KOG.
Functional classification by KEGG
KEGG is a database resource for understanding high-level functions and utilities of the biological system, such as the cell, the organism and the ecosystem, from molecular-level information, especially large-scale molecular data-sets, generated by genome sequencing and other high-throughput experimental technologies.[Citation25] To identify active biological pathways in I. rubescens, the unigenes were mapped to the reference canonical pathways in KEGG. As shown in , a total of 7359 unigenes were annotated into 258 different pathways. The mapped unigenes represented metabolism pathways of major biomolecules, such as carbohydrates, amino acids and lipids. The most prominent pathways were for the carbohydrate metabolism (736 members), translation (640 members), signal transduction (584 members) and folding, sorting and degradation (546 members). Interestingly, 279 unigenes were involved in the biosynthesis of other secondary metabolites (). Among them, the cluster for phenylpropanoid biosynthesis [PATH: ko00940] represented the largest group (108 members), followed by the flavonoid biosynthesis (47 members) [PATH: ko00941] and stilbenoid, diarylheptanoid and gingerol biosynthesis (40 members) [PATH: ko00945].
Figure 5. Functional classifiation of I. rubescens unigenes according to KEGG.
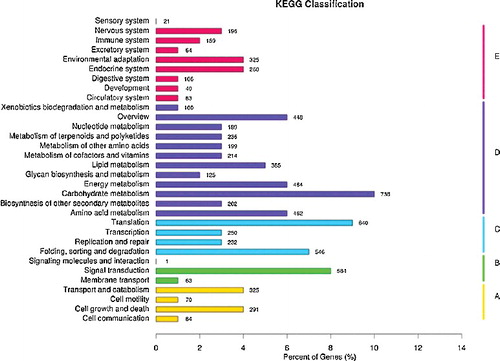
Figure 6. Unigenes related to other secondary metabolites.
Based on the above results, we found that many unigenes were assigned a putative gene or protein name descriptions and were categorized with GO terms and metabolic pathways. This will help us to better understand the gene expression profiles in I. rubescens. These annotations will provide a valuable resource for investigating specific processes, functions and pathways and will contribute to the identification of genes, which are involved in the pathways of secondary metabolite biosynthesis.[Citation26]
Based on the three public protein databases, we acquired a total of 36,921 CDSs, 25,101 CDSs predicted by BLASTX and 11,820 predicted by ESTScan, among which 826 were between 1000 and 2000 bp, 177 were over 2000 bp and 4903 were over 500 bp (). Unigenes were not identified as CDSs were likely non-coding RNAs.
Figure 7. Length distribution of CDS predicted by BLAST (a) and ESTScan (b)
Candidate genes encoding enzymes involved in terpene biosynthesis
Previous studies have shown that I. rubescens is rich in terpenes.[Citation27] Regulation of terpene biosynthesis is well characterized in other plants, such as Panax ginseng, Salvia miltiorrhiza, Artemisia annua and so on.[Citation28–30] But little is known about terpenes in I. rubescens, so we investigated the genes that control terpene biosynthesis. As shown in , we found 7 genes encoding enzymes involved in monoterpenoid biosynthesis; 80 genes were associated with terpenoid backbone biosynthesis, 35 unigenes were annotated as genes encoding diterpenoid biosynthesis and 8 unigenes were annotated into triterpenoid biosynthesis. It is known that terpene is derived from mevalonate and the plastidial 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway.[Citation31,Citation32] These two pathways contain a number of genes such as acetyl-CoA C-acetyltransferase, hydroxymethylglutaryl-CoA synthase, 1-deoxy-D-xylulose-5-phospate synthase, 1-deoxy- D-xylulose-5-phosphate reductoisomerase, farnesyl diphosphate synthase, ent-kaurene synthase and so on. Many studies have shown that diterpenoid derives from the MEP pathway.[Citation33] While the conversion of 1-deoxy-D-xylulose-5-phosphate to isopentenyl pyrophosphate, to geranylgeranyl pyrophosphate have been widely studied in many plant species, the next steps about diterpenoid are still unclear, especially the late steps of the pathway. So our study will be necessary for a full understanding of the pathway for terpene biosynthesis in I. rubescens and especially that of diterpenoid. This result will help us further understand the diterpenoid biosynthetic mechanisms and increase their level of accumulation by overexpressing these genes in I. rubescens.
Table 2. Transcripts involved in terpene biosynthesis in I. rubescens.
Expression patterns of five unigenes related to diterpenoid biosynthesis
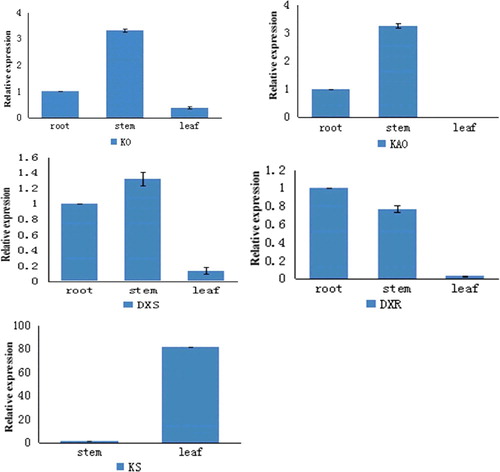
The candidate genes DXR, DXS, KS, KO, KAO were selected for further analysis and their expression patterns in roots, leaves and stems were analysed by qRT-PCR. The expression patterns of these genes are shown in . The highest expression of KO, DXS and KAO was detected in stems, where their expression was approximately 3.3, 1.32, 3.25-fold higher than that in roots, respectively and 8.96, 9.64-fold higher (for KO and DXS, respectively) than that in leaves, whereas KAO had no expression in leaves. The highest level of KS expression was observed in leaves and there was almost no expression in roots and stems. The highest expression of DXR was detected in roots. There was almost no expression in leaves.
Figure 8. Relative gene expression levels of different genes in tissues of I. rubescens.
I. rubescens has potential as a cancer cure.[Citation34] Studies on I. rubescens to date have mainly focused on the separation of chemical components, pharmacological action and tissue culture. Many studies indicate that the de novo transcriptome sequencing is an important technology in discovering secondary metabolite biosynthesis genes for medicinal plants.[Citation35–41] The lack of genomic data has hampered the research on the medicinal potential of I. rubescens. With the development of sequencing technology, many non-model plants, such as Codonopsis pilosula,[Citation42] Allium sativum [Citation43] and Siraitiagrosven-orii,[Citation44] have had their complete genomes sequenced. As genome resources for I. rubescens were not available, we used the RNA-seq to profile the transcriptome based on the Illumina sequencing platform and de novo analysis, from which we got a full understanding of I. rubescens transcriptome.
Our study focused mainly on terpene synthesis, especially diterpenoid biosynthesis, which has been used to treat esophageal cancer. Based on the analysis of the Illumina RNA-seq techonology, many genes encoding enzymes involved in terpene biosynthesis were identified. I. rubescens could represent a suitable model herbal plant for investigation of the terpenoid biosynthesis pathway and may likely facilitate functional studies aiming to produce larger quantities of this compound.
The large number of transcript sequences obtained in this study not only enriched the genomic resources for this species, but also benefited the researches on genetics, functional genomics and gene expression. This is the first exploration of the I. rubescens through the analysis of massive transcript data and the information will provide a basis for future studies on molecular biology, molecular breeding, physiology and biochemistry of this important species.
Conclusions
This study is the first report of I. rubescens transcriptome analysis. By using the Illumina platform, we investigated the transcriptome profile of I. rubescens. From the transcriptome, we obtained 44,626 unigenes, of which 25,678 unigenes were annotated from Nr, Nt, KO, Swiss-Prot, Pfam, GO and KOG databases. In addition, we discovered 35 valuable gene candidates for diterpenoid biosynthesis. These findings will provide a valuable database for further research on this species.
Disclosure statement
All authors declare that they have no conflict of interest.
Additional information
Funding
Related Research Data
References
- Liu J, Liang JY, Xie T. 冬凌草研究进展 [Development of Rabdosia rubescens (Hemsl.) Hara]. Strait Pharm J. 2004;16:1–7. Chinese.
- Li Q, Feng WS. 冬凌草化学成分、药理作用及开发研究进展 [The latest development of researches on the chemical constituents and pharmacological activity of Rabdosia rubescens (Hemls.) Hara]. J Henna Univ Chin Med. 2003;18:31–33. Chinese.
- Feng Y, Liang JY, Jing L. 冬凌草化学成分的研究 [Study on the constituents of Rabdosia rubescens Hemsl]. J China Pharm Univ. 2003;34:302–304. Chinese.
- Zheng XK, Li Q, Feng WS. 冬凌草中酚酸类化学成分研究 [Studies on chemical constituents of phenolic acids in Rabdosia rubescens]. Chin pharm J. 2004;39:335–336. Chinese.
- Han J, Ye M, Chen HB, et al. Determination of diterpenoids and flavonoids in Isodon rubescens by LC-ESI-MS-MS. Chromatographia. 2005;62:203–207. Chinese.
- Zuo HJ, Li D, Wu B, et al. 冬凌草的化学成分及其抗肿瘤活性 [Studies on the constituents of Rabdosia rubescens (Hemsl.) Hara and their antitumor activities in vitro]. J Shen yang Pharm Univ. 2005;22:258–262. Chinese.
- Yan XB, Lei M, Yu KE, et al. 冬凌草的化学成分研究 [Study on the constituents of Rabdosia rubescens]. Chem Res. 2006;17:80–82. Chinese.
- Liu X, Zhan R, Wang WG, et al. Three new 11,20-epoxy-ent-kauranoids from Isodon rubescens. Arch Pharm Res. 2012;35:2147–2151.
- Jia JL, Xin YW, Hui L, et al. 冬凌草甲素对NB4细胞的生长抑制作用及作用机制 [Inhibitory effect of oridonin on the proliferation of NB4 cells and its mechanism volume]. Chin-Ger J Clin Oncol. 2004;1:51–54. Chinese.
- Chen S, Liu R, Zhang HD. Efficacy of Rabdosia rubescens in the treatment of gingivitis. J Huazhong Univ Sci Technol Med Sci. 2009;5:659–663.
- Salminen A, Lehtonen M, Suuronen T, et al. Terpenoids: natural inhibitors of NF-κB signaling with anti-inflammatory and anticancer potential. Cell Mol Life Sci. 2008;65:2979–2999.
- Li YJ, Wang TX, Yang XF, et al. 冬凌草愈伤组织诱导及细胞培养的研究 [Studies on the induction of calli and cell culture of Rabdosia rubescens]. Chin Tradit Herb Drug. 2000;31:938–941. Chinese.
- Li DJ, Wei JF, Xu N, et al. 植物生长物质对冬凌草愈伤组织生长及褐化的影响 [Effect of plant growth substance on callus growth and browning of Rabdosia rubescens]. J Anhui Agric Sci. 2006;34:1118–1136. Chinese.
- Su XH, Dong CM, Wang CL. 冬凌草离体培养体系的建立及主要次生代谢产物的测定 [Establishment of culture system of Rabdosia rubescens (Hemsl.) Hara and content of the main secondary metabolites in its regenerated plantlets]. Acta Bot Boreal-Occid Sin. 2008;28:310–316. Chinese.
- Su XH, Dong CM, Wang WW. 氮碳源对冬凌草再生植株生长及次生代谢产物的影响 [Effects of sucrose concentration, nitrogen on the growth and main secondary metabolites accumulated of regeneration plant of Rabdosia rubescens (Hemsi.) Hara]. Acta Bot Boreal-Occid Sin. 2009;29:494–498. Chinese.
- Li GM, Li ZC, Xu KY, et al. 抗癌植物冬凌草种质资源遗传多样性分析 [Analysis on genetic diversity of germplasm resources of anticancer plant Rabdosia rubescens (Labiatae)]. J Trop Subtrop Bot. 2008;16:116–122. Chinese.
- Ai PF, Lu LP, Song JJ. Cryopreservation of in vitro-grown shoot-tips of Rabdosia rubescens by encapsulation-dehydration and evaluation of their genetic stability. PCTOC. 2012;108:381–387.
- Bleeker PM, Spyropoulou EA, Diergaarde PJ, et al. RNA-seq discovery, functional characterization, and comparison of sesquiterpene synthases from Solanum lycopersicum and Solanum habrochaites trichomes. Plant Mol Biol. 2011;77:323–336.
- Li P, Deng WQ, Li TH, et al. Illumina-based de novo transcriptome sequencing and analysis of Amanita exitialis basidiocarps. Gene. 2013;532(1):63–71.
- Liang C, Liu X, Yiu SM, et al. De novo assembly and characterization of Camelina sativa transcriptome by paired-end sequencing. BMC Genomics. 2013;14:146–156.
- Mudalkar S, Golla R, Ghatty S, et al. De novo transcriptome analysis of an imminent biofuel crop, Camelina sativa L. using Illumina GAIIX sequencing platform and identification of SSR markers. Plant Mol Bio. 2014;84:159–171.
- Garcia-Seco D, Zhang Y, Gutierrez-Mañero FJ, et al. RNA-Seq analysis and transcriptome assembly for blackberry (Rubus sp.Var. Lochness) fruit. BMC Genomics. 2015;16:5–15.
- Stone JD, Storchova H. The application of RNA-seq to the comprehensive analysis of plant mitochondrial transcriptomes. Mol Genet Genomics. 2015;290:1–9.
- Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652.
- Kyoto Encyclopedia of Genes and Genomes [Internet]. Kanehisa Laboratories; c1995–c2016 [ cited 2016 Oct 16]. Available from: http://www.genome.jp/kegg/.
- Huang Y, Wu X, Jian D, et al. De novo transcriptome analysis of a medicinal fungi Phellinus linteus and identification of SSR markers. Biotechnol Biotechnol Equip. 2015;29:395–403.
- Chen SQ, Song J, Cui C. 冬凌草二萜类成分的化学指纹图谱研究及评价[Research and evaluation on chemical fingerprints of diterpenoids from Isodon rubescens]. J Wuhan Bot Res. 2012;30:519–527. Chinese.
- Zhang R, Zhang BL, Gu-Cai L, et al. Enhancement of ginsenoside Rg1 in Panax ginseng hairy root by overexpressing the α-l-rhamnosidase gene from Bifidobacterium breve. Biotechnol Lett. 2015;37:2091–2096.
- Shi M, Luo XQ, Guan HJ, et al. Increased accumulation of the cardio-cerebrovascular disease treatment drug tanshinone in Salvia miltiorrhiza hairy roots by the enzymes 3-hydroxy-3-methylglutaryl CoA reductase and 1-deoxy-d-xylulose 5-phosphate reductoisomerase. Funct Integr Genomics. 2014;14:603–615.
- Yang JF, Adhikari MN, Liu H, et al. Characterization and functional analysis of the genes encoding1-deoxy- D -xylulose-5-phosphate reductoisomerase and 1-deoxy- D -xylulose-5-phosphate synthase, the two enzymes in the MEP pathway, from Amomum villosum Lour. Mol Biol Rep. 2012;39:8287–8296.
- Shen Q, Yan TX, Fu XQ, et al. Transcriptional regulation of artemisinin biosynthesis in Artemisia annua L. Sci Bull. 2016;19:1–8.
- Yang L, Yang CQ, Li CY, et al. Recent advances in biosynthesis of bioactive compounds in traditional Chinese medicinal plants. Sci Bull. 2015;1–15.
- Oldfield E, Lin FY. Terpene biosynthesis: modularity rules. Angew Chem Int Ed. 2012;51:1124–1137.
- Zhang WJ, Huang QL, Hua ZC. Oridonin: a promising anticancer drug from China. Front Biol. 2010;5:540–545.
- Hua WP, Zhang Y, Song J, et al. De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics. 2011;98:272–279.
- Hyun TK, Rim Y, Jang HJ, et al. De novo transcriptome sequencing of Momordica cochinchinensis to identify genes involved in the carotenoid biosynthesis. Plant Mol Biol. 2012;79:413–427.
- Zheng KY, Zhang GH, Jiang NH, et al. Analysis of the transcriptome of Marsdenia tenacissima discovers putative polyoxypregnane glycoside biosynthetic genes and genetic markers. Genomics. 2014;104:186–193
- Senthil K, Jayakodi M, Thirugnanasambantham P, et al. Transcriptome analysis reveals in vitro cultured Withania somnifera leaf and root tissues as a promising source for targeted withanolide biosynthesis. BMC Genomics. 2015;16:14–29.
- Zhang MF, Jiang LM, Zhang DM, et al. De novo transcriptome characterization of Lilium ‘Sorbonne’ and key enzymes related to the flavonoid biosynthesis. Mol Genet Genomics. 2015;290:399–412.
- Zhang SW, Ding F, He XH, et al. Characterization of the ‘Xiangshui’ lemon transcriptome by de novo assembly to discover genes associated with self-incompatibility. Mol Genet Genomics. 2015;290:365–375.
- Wang X, Li ST, Li J, et al. De novo transcriptome sequencing in Pueraria lobata to identify putative genes involved in isoflavones biosynthesis. Plant Cell Rep. 2015;34:733–743.
- Wang D, Cao LY, Gao JP. 党参转录组中SSR位点信息分析.中草药 [Data mining of simple sequence repeats in Codonopsis pilosula transcriptome]. Chin Tradit. Her Drugs. 2014;45:2390–2393. Chinese.
- Sun XD, Zhou SM, Meng FL, et al. De novo assembly and characterization of the garlic (Allium sativum) bud transcriptome by Illumina sequencing. Plant Cell Rep. 2012;31:1823–1828.
- Tang Q, Ma XJ, Mo CM, et al. An efficient approach to finding Siraitiagrosvenorii triterpene biosynthetic genes by RNA-seq and digital gene expression analysis. BMC Genomics. 2011;12:343.