
ABSTRACT
Site-directed mutagenesis (SDM) is a useful tool to study the functions of regulatory sequences of DNA and RNA, and the structures and functions of proteins. Numerous methods have been developed for either single or multiple SDM (MSDM). However, MSDM is sometimes difficult. Here we demonstrated that T4 DNA polymerase greatly enhanced the efficiency of MSDM. Moreover, we have also showed that it is efficient to clone multiple specific mutation-containing sequences simultaneously.
Introduction
Site-directed mutagenesis (SDM) is widely used for studying the regulatory DNA and RNA sequence functions, and protein structure and functions. Numerous strategies for single and multiple SDM (MSDM) have been reported.[Citation1–5] Among these strategies, the QuikChange® methods developed by Stratagene company are most commonly used to mutate the plasmid constructs. However, the original QuikChange® method for MSDM is inefficient. The primer design strategies for QuikChange® were improved by using partially overlapping primers instead of completely overlapping ones, resulting in exponential polymerase chain reaction (PCR) amplification and thus, greatly enhanced efficiency for both single and multiple SDM.[Citation6,Citation7] Nevertheless, for the application of the QuikChange method, it is a prerequisite that the wild-type targets of interest are first cloned into a plasmid. Recently, enzymes with exonuclease activity were used for SDM,[Citation8–11] which can mediate the complementary annealing of overlap sequences.[Citation11–15] These exonuclease-activity based methods are somewhat like the overlap extension PCR method. Extended primers are used to introduce overlap sequences into PCR products. The PCR product can be used as a megaprimer for overlap extension PCR, which allows gene-fusion or inserting of a large fragment into a plasmid.[Citation2,Citation5] The exonuclease activity-based methods use the exonuclease treatment to produce a single-stranded region of DNA ends, which allows the complementary annealing of overlap sequences.[Citation11–15] Interestingly, recent studies showed that QuikChange® and its improved methods indeed generate linear DNA molecules with homologous ends,[Citation11,Citation16] and our previous study showed that these homologous ends can be treated with exonuclease for complementary annealing of the overlapping DNA ends to enhance the efficiency of single SDM.[Citation11]
In an attempt to find a more convenient and efficient method for MSDM, we compared several different types of MSDM procedures. Here we described that T4 DNA polymerase can be used for MSDM, either for cloned plasmid constructs, or during the cloning procedure. For the former one, PCR can be used in a single-tube reaction to amplify mutated DNA exponentially, which allows fast mutagenesis without plasmid preparation and DpnI digestion. And for the latter one, DNA fragments with or without mutation can be simultaneously cloned.
Materials and methods
Comparison of different MSDM procedures
The pGL3-Olig2 plasmid was constructed by amplifying the 2.6-kb upstream sequence from the transcription start site of the mouse Olig2 gene, using the primers Olig2F and Olig2R (), and cloned into the NheI and XhoI site of the pGL3-basic vector (Promega Biotech Co. Ltd., Beijing, China) using the method previously described by us.[Citation11] The QuikChange method was performed according to the manufacturer's instructions of QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Stratagene, now a part of Agilent Technologies, La Jolla, CA, USA), using 100 ng of pGL3-Olig2 plasmid as a template, and Olig2MF1 and Olig2MF2 as primers. The QuikChange reaction product was treated with DpnI at 37 °C for more than 1 h to deplete the original pGL3-Olig2 plasmid. For the improved MSDM, 1 µL of pGL3-Olig2 plasmid-containing bacterial culture or 1 pg of pGL3-Olig2 plasmid was used as a template, and Olig2MF1, Olig2MR1, Olig2MF2 and Olig2MR2 were used as primers. PrimeSTAR GXL DNA polymerase (Takara Biotech Co. Ltd., Dalian, China) was used for PCR, and PCR was performed under the following cycling conditions: initial denaturation at 98 °C for 30 s; followed by 25 cycles of denaturation at 98 °C for 10 s, and annealing and extension at 66 °C for 3 min 30 s; and a final extension at 66 °C for 5 min. The PCR product was purified by DNA Purification Kit (CWBio Co. Ltd., Beijing, China). And 100 ng of the purified PCR product was treated by 1 U of T4 DNA polymerase (Fermentas, now a part of Thermo Fisher Scientific Inc., Waltham, MA, USA) or T7 exonuclease (New England Biolabs, Ipswich, MA, USA) slightly modified from our previous method.[Citation11] The enzyme treatments were conducted in a MyCycler™ thermo-cycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA) using the following program: incubate at 20 °C for 3 min, followed by 75 °C for 5 min and 48 °C for 10 min. The enzyme-treated products were then transformed into competent DH5α cells (CWBio Co. Ltd., Beijing, China) according to the user manual, and one fifth of the transformation suspension was spread onto lysogeny broth (LB) agar plates with ampicillin for colony formation. Picked colonies were sent to Shanghai Sunny Biotechnology Co. Ltd., (Shanghai, China) for sequencing.
Table 1. PCR primers used in this study.
Simultaneous cloning of multiple fragments with mutations
Three fragments of the 2.6 kb upstream sequence of the mouse Olig2 gene were separately PCR amplified by primer pairs Olig2F and Olig2MR1, Olig2MF1 and Olig2MR2 and Olig2MF2 and Olig2R. The PCR products were purified by DNA Purification Kit (CWBio Co. Ltd., Beijing, China). The molecular weights of the three fragments were 169 bp, 1928 bp and 590 bp, respectively. Plasmid pGL3-basic (Promega) was cut by restriction enzymes NheI and XhoI (New England Biolabs, Ipswich, MA, USA), and then the enzymes were heat inactivated by incubating at 80 °C for 20 min. After that, 50 ng of enzyme-cut pGL3-basic plasmid, 30 ng of the 169 bp fragment, 100 ng of the 1928 bp fragment and 50 ng of the 590 bp fragment were mixed together, and treated by 1 U of T4 DNA polymerase at 20 °C for 3 min, followed by 75 °C for 5 min and 48 °C for 10 min. The colonies were randomly picked by pipette tips and transferred into 1 ml LB medium with ampicillin, and 1 μL of the medium was withdrawn for subsequent PCR verification. PCR was performed using the primers Olig2F and Olig2R under the following programme: initial denaturation at 98 °C for 30 s; followed by 30 cycles of denaturation at 98 °C for 10 s, and annealing and extension at 66 °C for 1 min 30 s; and a final extension at 66 °C for 5 min. Five microlitres of the PCR products was used for electrophoresis in 1.5% agarose gel containing DNAGREEN (UV) (Tiandz, Inc., Beijing, China). A DL10,000 DNA Marker (Takara Biotech Co. Ltd., Dalian, China) was used as a molecular weight standard. Gel images were captured by ChemiDoc™ Imaging Systems (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Data analysis
Results from three independent experiments were used for statistical analysis. One-tailed student's t-test in Microsoft Excel was used for statistical analysis.
Results and discussion
T4 DNA polymerase greatly improves MSDM
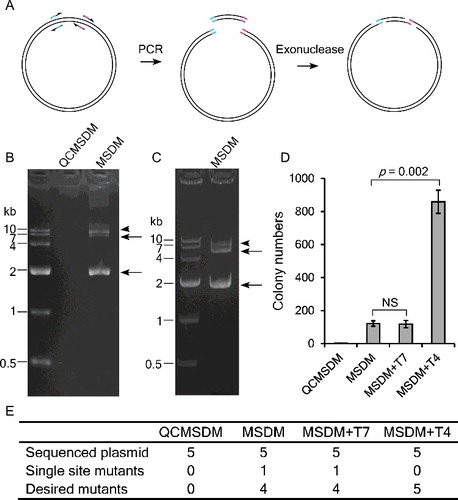
Our previous study showed that a treatment with T4 DNA polymerase, which is well known for its 3’→5’ exonuclease activity, greatly improves the efficiency of single SDM.[Citation11] As depicted in (A), we proposed that exonuclease treatment may also increase the efficiency of MSDM. To test this hypothesis, we first used a single-tube mutagenesis PCR reaction to amplify targets and compared it to the QuikChange Multi Site-Directed Mutagenesis method. Although we used as much as 100 ng of template plasmid DNA, there was no visible target band amplified using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit ((B)). In contrast, the improved method [Citation7] showed a robust amplification of targets from 0.5 μL of bacterial culture ((B)) or 1 pg of template plasmid DNA ((B)). The transformation results showed that the original QuikChange method is inefficient at producing multiple mutations, while the improved method [Citation7] generated more than 100 colonies, in which 4 of 5 randomly picked colonies contain the desired mutations ((B) and 1(C)). We next tested two enzymes, T4 DNA polymerase and T7 exonuclease, to see if they could increase the efficiency of MSDM. Treatment with T7 exonuclease did not increase the yield of colonies; however, treatment with T4 DNA polymerase further increased the colony yield approximately seven-fold ((B) and 1(C)).The results indicated that the exonuclease activity of T4 DNA polymerase but not of T7 exonuclease enhanced the homologous-end recombination.
Figure 1. Use of exonuclease to improve multiple site-directed mutagenesis (MSDM). (A) Partially overlapping primers are used to amplify mutated DNA fragments with homologous ends. Exonuclease treatment helps the complementary annealing of these homologous ends. (B) Mutagenesis reaction carried out by the QuikChange method (QCMSDM) or partially overlapping primers as depicted in (A), 100 ng and 1 pg of pGL3-Olig2 plasmid were, respectively, used as a template in the two reactions. The desired bands are indicated by arrows; an additional band is indicated by an arrowhead. (C) Mutagenesis reaction was performed using 1 μL of pGL3-Olig2-containing bacterial culture. (D) T4 DNA polymerase treatment increased the yield of colonies for mutagenesis. PCR products were treated with T7 exonuclease (T7) or T4 DNA polymerase (T4) before transformation. Colony numbers are shown as means and standard deviation of three independent experiments. (E) Sequencing verification of mutagenesis.
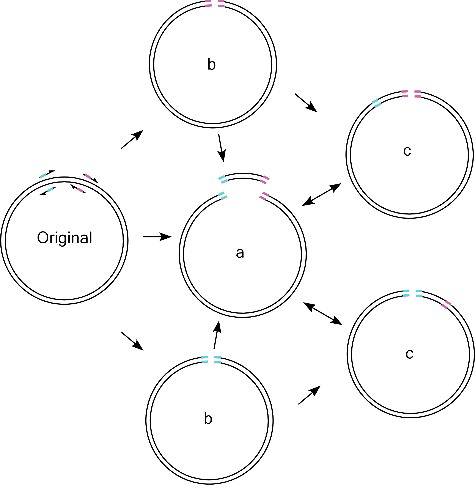
The PCR reaction produced three bands ((B)). The two shorter bands were those amplified by the primer pairs of Olig2MF1 and Olig2MR2, and Olig2MF2 and Olig2MR1 (), respectively. The longest band is indeed a mixture of different types of DNA fragments, including the direct and indirect amplification products by the primer pairs of Olig2F1 and Olig2R1, and Olig2F2 and Olig2R2 (). The direct amplification products carry a single site mutation, which should only account for a small amount of the longest band (). The indirect amplification products account for the major part of the longest band, which are primary overlap extension products using the shorter two fragments as megaprimers () and further amplified by the primer pairs of Olig2F1 and Olig2R1, and Olig2F2 and Olig2R2 ().
Figure 2. Products generated in MSDM PCR. During the PCR, the undesired single mutation products (b) can be used by primers and megaprimers to generate desired products (a and c), which in turn inhibits the amplification efficiency of single mutation products (b).
T4 DNA polymerase has an intrinsic proofreading activity and when the nucleotides (dNTPs) are insufficient, it degrades DNA in the 3’→5’ direction, whereas T7 exonuclease degrades DNA in the 5’→3’ direction. For mediating complementary annealing of overlapping sequences, T4 DNA polymerase requires removal of dNTPs from the PCR reaction, while T7 exonuclease does not. These different properties mean that T7 exonuclease may be a more convenient enzyme for MSDM. Unexpectedly, however, treatment with T7 exonuclease did not increase the efficiency of MSDM. The possible reason is that T7 exonuclease is resistant to heat inactivation and may over-digest the PCR products during complementary annealing.
MSDM during cloning procedure
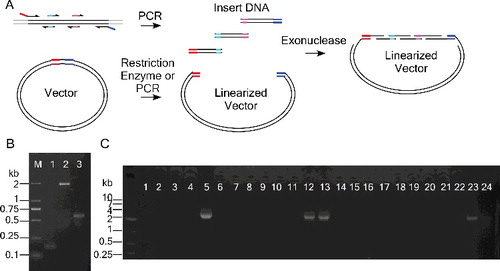
As in most cases, wild-type sequences were not cloned ready for mutagenesis; a strategy independent of cloned wild-type sequences is needed to accelerate the mutagenesis procedure. For this purpose, we adapted the use of T4 DNA polymerase to clone multiple sequences ((A)). We separately amplified the mutated fragments ((B)). Subsequently we mixed these fragments with a linearized vector for T4 DNA polymerase treatment, which enables simultaneously cloning of mutation-containing multiple sequences ((A)). Transformation of the T4 DNA polymerase-treated DNA yielded more than 1000 colonies. The colony PCR result showed that among the 24 randomly picked colonies, 4 contained the desired mutations ((C)). The results indicated that the wild-type sequence and the mutated sequence can be cloned simultaneously. Compared to other MSDM procedures,[Citation7–9] which require days of cloning and verification of the wild-type sequence prior to mutagenesis, our mutagenesis-coupled cloning strategy can further speed up the mutagenesis procedure.
Figure 3. Cloning coupled with mutagenesis. (A) Shorter mutation-containing fragments with overlap ends are amplified in parallel. The overlapping sequences can be annealed by exonuclease treatment. (B) PCR products amplified using the three primer pairs: Lane 1, Olig2F and Olig2MR1; Lane 2, Olig2MF1 and Olig2MR2; Lane 3, Olig2MF2 and Olig2R. (C) Colony PCR using the primers of Olig2F and Olig2R. Colonies 5, 12, 13 and 23 were verified to contain the desired sequence.
Conclusions
In summary, we have demonstrated that T4 DNA polymerase can be used to increase the efficiency of MSDM. Taking advantage of the exponential PCR amplification using partially overlapping primers, the procedures for plasmid extraction and DpnI treatment are omissible. Moreover, using the T4 DNA polymerase-mediated multiple sequence assembly, MSDM can be further accelerated by direct cloning of multiple sequences with mutations.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA. 1985;82:488–492.2.
- Ho SN, Hunt HD, Horton RM, et al. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59.1.
- Vandeyar MA, Weiner MP, Hutton CJ, et al. A simple and rapid method for the selection of oligodeoxynucleotide-directed mutants. Gene. 1988;65:129–133.1.
- Sarkar G, Sommer SS. The "megaprimer" method of site-directed mutagenesis. Biotechniques. 1990;8:404–407.
- Geiser M, Cebe R, Drewello D, et al. Integration of PCR fragments at any specific site within cloning vectors without the use of restriction enzymes and DNA ligase. Biotechniques. 2001;31:88,90,92.
- Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res [Internet]. 2004 [cited 2015 Oct 10];32:e115. Available from: http://dx.doi.org/10.1093/nar/gnh110
- Liu H, Naismith JH. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol [Internet]. 2008 [ cited 2015 Oct 10];8:91. Available from: http://dx.doi.org/10.1186/1472-6750-8-91
- Liang X, Peng L, Li K, et al. A method for multi-site-directed mutagenesis based on homologous recombination. Anal Biochem. 2012;427:99–101.1.
- Mitchell LA, Cai Y, Taylor M, et al. Multichange isothermal mutagenesis: a new strategy for multiple site-directed mutations in plasmid DNA. ACS Synth Biol. 2013;2:473–477.
- Taniguchi N, Nakayama S, Kawakami T, et al. Patch cloning method for multiple site-directed and saturation mutagenesis. BMC Biotechnol [Internet]. 2013 [ cited 2015 Oct 10];13:91. Available from: http://dx.doi.org/10.1186/1472-6750-13-91
- Sun S, Huang H, Qi YB, et al. Complementary annealing mediated by exonuclease: a method for seamless cloning and conditioning site-directed mutagenesis. Biotechnol Biotechnol Equip. 2015;29:105–110.
- Yang YS, Watson WJ, Tucker PW, et al. Construction of recombinant DNA by exonuclease recession. Nucleic Acids Res. 1993;21:1889–1893.8.
- Tseng H. DNA cloning without restriction enzyme and ligase. Biotechniques. 1999;27:1240–1244.
- Li MZ, Elledge SJ. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods. 2007;4:251–256.
- Gibson DG, Young L, Chuang RY, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345.
- Xia Y, Chu W, Qi Q, et al. New insights into the QuikChange™ process guide the use of Phusion DNA polymerase for site-directed mutagenesis. Nucleic Acids Res [Internet]. 2015 [cited 2015 Oct 10];43:e12. Available from: http://dx.doi.org/10.1093/nar/gku1189