
ABSTRACT
The aim of this study was to establish a system for high expression of Aspergillus niger glucose oxidase (GOD) with glyceraldehyde-3-phosphate dehydrogenase gene promoter (pGAP) in Pichia SMD1168. The GOD gene from A. niger accc30161 was inserted into pGAPZαA plasmid carrying a pGAP promoter and was expressed in yeast Pichia pastoris. The expression vector, pGAPZαA-GOD, was validated by colony polymerase chain reaction (PCR), agarose gel electrophoresis, restriction enzyme analysis and sequencing methods. The pGAPZαA-GOD vector was transformed into yeast P. pastoris SMD1168 by electroporation and the positive strain was validated by PCR. The expression and enzyme activity of the recombinant GOD were analysed by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and enzymatic detection. The recombinant GOD in yeast was purified by ion exchange chromatography, and its biochemical properties (dynamics, thermal and pH stability) were analysed. The obtained results showed that the expression vector, pGAPZαA-GOD, was successfully constructed to express A. niger accc30161 GOD protein. After transformation, the pGAPZαA-GOD DNA fragment had been integrated into the P. pastoris SMD1168-GOD genome. High expression of GOD was achieved in SMD1168-GOD, and the enzyme activity of GOD reached 107.18 U/mL in the supernatant of the culture medium at 30 °C and pH 6. The activity of recombinant GOD protein in yeast was 1.35-fold higher than the commercial A. niger GOD, and its affinity to glucose, thermal stability and pH stability are similar to commercial GOD. Using pGAP promoter, A. niger GOD protein is efficiently expressed in recombinant P. pastoris SMD1168-GOD with defective protease.
Introduction
Glucose oxidase (GOD; EC 1.1.3.4) is an aerobic dehydrogenase which is widely used in quantitative analysis, medical testing, food industry, biosensors and many other fields.[Citation1–3] Currently, GOD is mainly produced and purified from Aspergillus niger and Penicillium, but the yield is relatively low and the purification process is complicated.[Citation4–6] It is practical to build an excellent GOD-producing strain by genetic engineering. The yeast Pichia pastoris expression system is one of the most widely used eukaryotic expression systems due to its ability to highly express proteins without contamination of other products. Furthermore, P. pastoris has many advantages for strictly regulating expression, processing, folding and post-translational modification of exogenous proteins and secretion of exogenous proteins outside the extracellular membrane.[Citation7–9] Many scholars have reported optimized conditions for production of recombinant GOD enzyme by using the P. pastoris expression system.[Citation10–13] To the best of our knowledge, however, until now, there are no reports about expression of recombinant GOD with glyceraldehyde-3-phosphate dehydrogenase gene promoter (pGAP) in protease-deficient P. pastoris strain. In this study, we constructed the expression vector containing the GOD gene from A. niger accc30161 strain with pGAP promoter, and then transformed it to P. pastoris SMD1168. GOD protein was efficiently expressed in protease-deficient P. pastoris regulated by pGAP.
Materials and methods
Strains, vectors and reagents
A. niger accc30161 strain was kindly given by Professor Meixue Dai from Department of Biotechnology, College of Life Sciences, Shandong Normal University. P. pastoris SMD1168 and pGAPZαA plasmid were purchased from Invitrogen (Carlsbad CA, USA). Escherichia coli (E. coli). DH5α and 2×Easy Taq PCR SuperMix were purchased from TransGen Biotech (Beijing, China). Q Sepharose™ Fast Flow was purchased from Amersham Biosciences (Piscataway, NJ, USA). Restriction enzymes Not I, Kpn I and EcoR I were purchased from Takara (Dalian, China). T4 DNA ligase and restriction enzyme Avr II were purchased from Fermentas (Ontario, Canada). Primer synthesis and gene sequencing were completed by Shenzhen Genomics Company (Shenzhen, China). A. niger GOD and o-dianisidine were purchased from Sigma (St. Louis, MO, USA). Omega SP Fungal DNA kit, plasmid extraction kit and agarose gel extraction kit were purchased from Omega (Norcross, GA, USA). Bicinchoninic acid (BCA) protein assay kit was purchased from Nanjing KGI Biotechnology Co., Ltd (Nanjing, China). DH5α cells were cultured and transformed in low-salt Luria--Bertani (LB) medium (5 g/L yeast extract, 10 g/L tryptone, 4 g/L NaCl and 15 g/L agar). A. niger accc30161 was cultured in potato dextrose medium (200 g/L potato and 20 g/L dextrose). SMD1168 strain was cultured and transformed in yeast extract peptone dextrose (YPD) medium (10 g/L yeast extract, 20 g/L tryptone and 20 g/L glucose). Horseradish peroxidase was purchased from Thermo Scientific Pierce Co. Ltd. (Rockford, IL, USA).
Amplification of A. niger GOD DNA and construction of recombinant plasmid
The genomic DNA of A. niger accc30161 strain was extracted according to the protocol of the Omega SP Fungal DNA kit. The DNA fragment containing the GOD gene was amplified by polymerase chain reaction (PCR), in which the template was A. niger genomic DNA and the primers were P1 and P2 (). The PCR was performed in an Eppendorf AG 22331 MasterCycler (Eppendorf, Hamburg, Germany). The reaction system included: 2×Easy Taq PCR SuperMix 25 μL, 2 μL of template DNA, 2 μL of 5' primer and 2 μL of 3' primer and 19 μL of sterilized ddH2O. The cycle conditions were the following: 94 °C for 3 min, then 30 cycles at 94 °C for 30 s, 50 °C for 30 s and 70 °C for 2 min, followed by 70 °C for 10 min. The PCR products (more than 2 kb) were analysed by agarose gel electrophoresis (Beijing Liuyi DYY-8C electrophoresis apparatus, Beijing Liuyi Instrument Factory, Beijing, China) and sequenced.
Table 1. Sequences of primers used in this study.
The GOD gene fragment was also amplified by PCR using genomic DNA of A. niger accc30161 as a template. The forward primer (P3) and the reverse primer (P3) were listed in . The reaction system and the cycle conditions were the same as above. The amplified product was ligated into the pGAPZαA vector between the Kpn I and Not I sites to produce a pGAPZαA-GOD plasmid. The recombinant plasmid was transformed into E. coli DH5α and selected on low-salt LB medium with 25 µg/mL Zeocin™ (Invitrogen Corp., Carlsbad, CA, USA). It was confirmed that the GOD gene inserted into recombinants with right orientation by PCR amplification and EcoR I digestion. The primers for recombinant detection were pGAP and 3′AOX1 as shown in . And the PCR conditions were the same as earlier.
Transformation of Pichia cells
Avr II was used to linearize the pGAPZαA-GOD plasmid, and the linear DNA was transformed into competent Pichia SMD1168 cells by electro-transformation.[Citation14] The transformants were selected on YPD medium plates with 100 µg/mL Zeocin at 30 °C for 96 h. Positive colonies were randomly selected for further culture in YPD liquid medium (with 100 µg/mL Zeocin). The cells were collected after culturing for three days and the DNA was extracted by an Omega SP Fungal DNA kit. Then, the transformants were selected and validated by PCR amplification with the primers P3 and P4 (). The PCR conditions were the same as described earlier.
Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE)
The supernatant of the SMD1168-GOD culture medium was harvested at 1500× g for 10 min (Sigma 2-16K, Sigma Laborzentrifugen GmbH, Osterode am Harz, Germany). The concentration of protein was determined by BCA method. The supernatant was loaded on a 10% sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) gel for electrophoresis (Beijing Liuyi DYY-8C electrophoresis apparatus, Beijing Liuyi Instrument Factory, Beijing, China) and stained with Coomassie brilliant blue R-250. After decolouring the gel with destaining solution (methanol:acetic acid:water = 5:1:4), the band of GOD protein was investigated.
Detection of GOD enzymatic activity
The standard curve was made using fresh A. niger GOD (Sigma, St. Louis, MO, USA) as described in.[Citation15, Citation16] The GOD activity was detected based on the standard curve. Assays were performed in a volume of 3.2 mL in 2 mL glycerol, 1 mL glucose solution (2 mg/mL, which was formulated with 0.1 mol/L sodium citrate buffer, pH 5.6), 100 μL o-dianisidine solution (2 mg/mL, formulated with sterilized water) and 100 μL of horseradish peroxidase solution (100 U/mL, which formulated with 0.1 mol/L sodium citrate buffer, pH 5.6). The mixture was incubated at 30 °C for 10 min and was then added to 20 µL GOD standard solution or supernatant of SMD1168-GOD culture medium to initiate the reaction. The reaction was incubated at 30 °C for another 10 min and was quenched by addition of 2 mL of 5 mol/L HCl. The activity of GOD was measured at 520 nm with a microplate reader (Multiskan FC, Thermo Fisher Scientific Laboratory Equipment, Hudson NH, USA).
Optimization of SMD1168-GOD culture conditions
The activity of GOD protein was detected in the supernatant of SMD1168-GOD cultured in liquid YPD medium for five days at 20, 25, 30, 35 or 40 °C. The activity of GOD protein was also detected from the supernatant of SMD1168-GOD cultured in liquid YPD medium (pH 4, pH 5, pH 6, pH 7 or pH 8) for five days at 30 °C.
Purification of recombinant GOD protein
The cultured yeast cells were centrifuged at 4000× g for 5 min at room temperature (Avanti J-E Centrifuge, Beckman Coulter, Inc., Palo Alto, CA, USA). The supernatant was ultrafiltered at 4000× g for 20 min at room temperature using the Amicon Ultra 15 mL centrifugal filter with a membrane nominal molecular weight limit (NMWL) of 100 kDa (Millipore Corp., Billerica, MA, USA), followed by dialysis in 0.002 mol/L sodium citrate buffer, pH 6.0. The concentrated supernatant was loaded onto a column of Q Sepharose™ Fast Flow ion exchange chromatography that was already equilibrated with 0.01 mol/L sodium citrate buffer (pH 6.0). The fraction of active GOD protein was collected by elution with a single linear gradient of 0–1.0 mol/L sodium chloride salt buffer (pH 6.0) and then SDS-PAGE analysis was performed.
Kinetics, thermal stability and pH stability analysis of recombinant GOD protein
The enzyme kinetics of recombinant GOD were studied in the solution with oxygen saturation, pH 6.0 at 30 °C. The reaction rate of recombinant GOD and commercial GOD standard to substrate glucose with different concentration (10, 12, 14, 16, 18, 20, 25, 30, 35, 40, 50, 60, 80 and 100 mmol/L) was measured. The kinetic constants of two GOD proteins were measured by double reciprocal plot method.
The thermal stability of recombinant GOD protein was measured through comparing the residual activity of the 15 U/mL (pH 6.0) recombinant GOD and commercial GOD standards by incubating for 1 h at different temperatures: 10, 20, 30, 40, 50, 60 or 70 °C.
The pH stability of recombinant GOD protein was studied by using a buffer of different pH values (pH 2, pH 3, pH 4, pH 5, pH 6 or pH 7, in sodium citrate buffer; pH 8, pH 9 and pH 10 in Tris-HCl buffer) to dilute recombinant GOD and commercial GOD to 15 U/mL. Then, the activity of GOD was measured after incubation at 30 °C for 12 h.
Data analysis
The SPSS 17.0 software was used for statistical analysis. All experiments were repeated three times and the data are shown as the mean values with standard deviation (±SD). Statistically significant differences were determined based on two-tailed Student's t-test. Values of P < 0.05 were regarded as indicating statistically significant differences.
Results and discussion
The main proteases encoded in the P. pastoris genome include protease A (aspartic proteases), protease B (cysteine proteolytic enzymes) and carboxypeptidase, all of which are acidic proteases. Under the optimum pH for yeast growth, these three enzymes retain high activity, which would decrease the production of target protein. In this study, we adopted the protease A (pep4) and protease B (prb1) deficient strain P. pastoris SMD1168, which could highly decrease or eliminate the effect of protease activity. Through slowing down the proteolysis, the production of target protein will be increased. Several studies have shown that the protease-deficient strain SMD1168 could highly express lipoxygenase,[Citation17] lysozyme LYZL6,[Citation18] glucosamine-6-phosphate synthase,[Citation19] streptokinase [Citation20] and other exogenous proteins.
The promoter is the key element to regulate target gene expression. The commonly used promoters in the P. pastoris expression system include the alcohol oxidase gene promoter pAOX1, pGAP, 3-phosphoglycerate dehydrogenase gene promoter pScgpd, glutathione-dependent formaldehyde dehydrogenase gene promoter pFLD1, peroxisomal matrix protein gene promoter pPEX8, guanosine triphosphate-binding protein gene promoter pYPT1, etc.[Citation21–23] Among them, pAOX1 is the most widely used promoter in yeast, as it is a strong, methanol inducible promoter for efficient expression of exogenous protein. However, methanol is easily flammable and toxic. The application of methanol in the field of food and medicine is severely restricted. Another strong constitutive promoter is pGAP. The pGAP promoter is inserted into the P. pastoris genome along with the target gene by homologous recombination, which ensures the genetic stability of exogenous genes in the yeast genome and stable expression of the target protein even after multiple passages. It has been demonstrated that the amount of exogenous protein is equal to or greater than that using the pAOX1 promoter when using the pGAP promoter.[Citation24] And the pGAP promoter is a non-alcohol inducible promoter, by which the expression system does not involve methanol. The fermentation process is simple and the production has no methanol contamination.[Citation25] Therefore, construction of the P. pastoris expression system by using the pGAP promoter attracts more attention. Until now, angiostatin,[Citation26] thermophilic actinomycetes α-amylase,[Citation27] β-mannanase,[Citation28] laccase,[Citation29] barley α-amylase,[Citation30] lipase LIP2 [Citation24] and β-galactosidase,[Citation31] etc. have been expressed under the promoter of pGAP.
Cloning and analysis of A. niger accc30161 GOD sequence
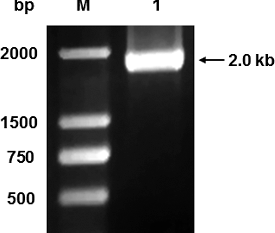
To amplify the DNA sequence of the GOD gene, we applied PCR methods. With A. niger accc30161 genomic DNA as the template and P1 and P2 as primers, a fragment of about 2.0 kb containing the GOD gene was amplified (). The DNA fragment has 1818 bp without introns and encodes 605 amino acids. The amplified sequence was consistent with the known GOD gene sequences like A. niger ATCC9029 GOD by blasting. The results indicate that A. niger strain accc30161 also contains the GOD gene and the sequence is the same as that of other A. niger GOD genes.
Figure 1. Agarose gel electrophoresis of PCR amplification product from accc30161. M: DNA Marker DL2 000 (Takara Biotechnology Co., Ltd., Dalian, China); 1: PCR amplification products.
Construction of recombinant pGAPZαA-GOD plasmid
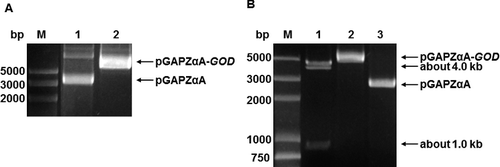
The plasmid pGAPZαA is a secreted expression vector, which carries its own pGAP promoter to constitutively express foreign proteins in P. pastoris. The sequence of the GOD gene was amplified by P3 and P4 primers. The PCR product was purified and inserted into the pGAPZαA vector to construct a recombinant expression vector pGAPZαA-GOD. This plasmid was transformed into E. coli DH5α and was selected by LB medium with Zeocin and low salt. Colony PCR was done with primers pGAP and 3′AOX1. In total, 18 out of 23 colonies that were selected randomly amplified the expected band (2.3 kb) of the GOD gene. We isolated the plasmid from ‘correct’ colonies and confirmed them by agarose gel electrophoresis. As shown in ((A)), the size of the pGAPZαA vector was 3.1 kb, while the size of the correct pGAPZαA-GOD vector was 4.9 kb. The ‘correct’ colonies were further confirmed by EcoR I digestion, and two bands of 4.0 and 1.0 kb DNA fragments were also detected, which was consistent with the expected result ((B)). Through sequencing the DNA of the recombinant vector, it was confirmed that the reading frame of the target gene had been correctly inserted downstream of the α-factor signal of pGAPZαA vector. All these results showed that the P. pastoris expression vector, pGAPZαA-GOD, was detected to express GOD of A. niger accc30161.
Figure 2. Identification of the recombinant plasmid pGAPZαA-GOD by agarose gel electrophoresis and its restriction digestion. Identification (A) of the recombinant plasmid pGAPZαA-GOD: lane M, DNA Marker DL5 000 (Takara Biotechnology Co., Ltd., Dalian, China); lane 1, pGAPZαA; lane 2, pGAPZαA-GOD. Restriction digestion (B) of the recombinant plasmid pGAPZαA-GOD with EcoR I: lane M, DNA Marker DL5 000; lane 1, digestion products of the plasmid pGAPZαA-GOD by EcoR I; lane 2, pGAPZαA-GOD plasmid; lane 3, pGAPZαA plasmid.
Expression and enzyme activity analysis of yeast recombinant GOD protein
After transforming P. pastoris with the pGAPZαA-GOD plasmid, we selected the ‘correct’ colonies by PCR with P3 and P4 primers and the genomic DNA of P. pastoris SMD1168-GOD and its control strain SMD1168. The agarose gel electrophoresis results showed that a band of 2.0 kb DNA fragment was obtained from the SMD1168-GOD sample, while no visible band was obtained from its control strain ((A)). This result indicated that the GOD gene of A. niger accc30161 had been successfully inserted into the genome of P. pastoris.
Figure 3. Verification of accc30161 GOD gene from SMD1168-GOD by PCR amplification and identification of GOD protein in the culture supernatant of SMD1168-GOD by SDS-PAGE analysis. PCR amplification analysis (A) of accc30161 GOD gene from SMD1168-GOD: M, DNA Marker DL2 000; 1, SMD1168-GOD; 2, SMD1168. SDS-PAGE analysis (B) of proteins in the culture supernatant of SMD1168-GOD: M, protein size marker (Fermentas, Burlington, Ontario, Canada); 1, SMD1168; 2, SMD1168-GOD.
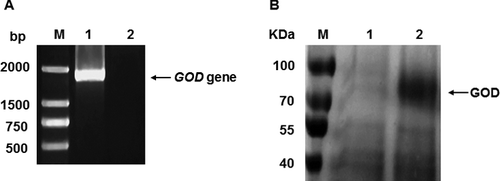
The supernatant of the SMD1168-GOD culture medium was loaded onto SDS-PAGE for further analysis. As shown in ((B)), a predominant band at approximately 70 kDa was observed. This protein size is similar to the size of the GOD protein of A. niger, which is 65 kDa. This suggested that the GOD protein of A. niger was expressed in the SMD1168-GOD strain. We also detected the enzyme activity in the supernatant of the SMD1168-GOD strain, which also suggested that the recombinant GOD protein was active.
Optimization of recombinant yeast culture conditions
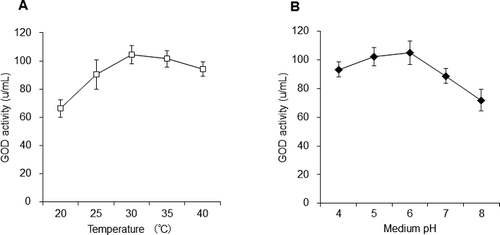
In order to obtain the best conditions for expressing the GOD protein in P. pastoris, we investigated different temperatures and different pH values for growth of P. pastoris. The results showed that temperature had apparent influence on GOD activity. And at 30 °C, the supernatant of the SMD1168-GOD medium showed the highest enzyme activity ((A)). The pH of the culture medium also had a significant effect on GOD activity ((B)). The supernatant of the SMD1168-GOD medium had highest enzyme activity at 30 °C and pH 6.
Figure 4. Effect of temperature (A) and medium pH (B) on the expression of the GOD gene in SMD1168-GOD strain.
Purification of recombinant yeast GOD protein
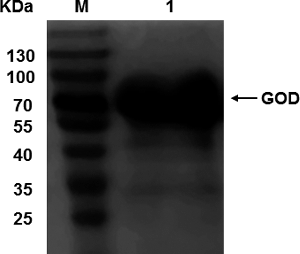
The GOD expressed in P. pastoris was purified by Q Sepharose™ Fast Flow ion exchange chromatography. The purified GOD protein was analysed using SDS-PAGE method, and the result showed that a thick band at approximately 70 kDa was observed (). The enzymatic activity of GOD was compared with that of commercial GOD. The activity of the recombinant GOD (537.44 U/mg) was 1.35 times stronger than that of the commercial product (398.10 U/mg).
Figure 5. SDS-PAGE analysis of purified recombinant GOD protein expressed by SMD1168-GOD strain. M: Protein size marker (Fermentas, Burlington, Ontario, Canada); 1: purified recombinant GOD protein.
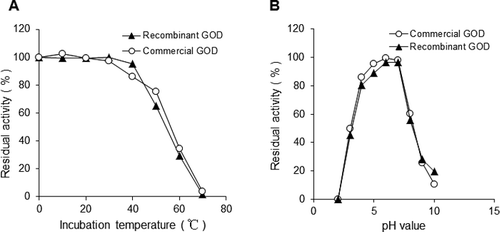
Kinetics, thermal stability and pH stability of recombinant GOD protein
We analysed the enzyme kinetics of this recombinant GOD protein and the commercial GOD. It was found that these two proteins had similar affinity to glucose. The Km value of the recombinant GOD was 45.06 mmol/L, while that of the commercial GOD was 39.76 mmol/L.
The thermal stability and pH stability of recombinant GOD and commercial GOD were also analysed. They were all stable at 4–40 °C, pH 4–7. When the temperature was above 40 °C, the activity of the two enzymes began to decrease. When the temperature reached 70 °C, both enzymes were almost completely deactivated ((A)). The activities of these two enzymes were also decreased when the pH was lower than 4 or more than 7 ((B)).
Figure 6. Thermal stability (A) and pH stability (B) of recombinant and A. niger GOD protein under the same conditions.
Previous studies show that the activity of GOD protein from A. niger ATCC9029 under usage of the pGAP promoter was only 0.83 and 1.62 U/mL after culturing recombinant P. pastoris for 48 or 240 h, respectively.[Citation32] The A. niger Z-25 GOD activity was 40 U/mL when the recombinant protein was expressed in P. pastoris SMD1168 strain using the pAOX1 promoter.[Citation10] To the best of our knowledge, the highest reported activity of wild A. niger NRLL-3 GOD in the culture medium was 17.5 U/mL when it was expressed in the P. pastoris KM71H strain with pAOX1 as a promoter.[Citation12] Using the pAOX1 promoter to express GOD in the P. pastoris GS115 strain, the activity of GOD in culture medium was 99 U/mL.[Citation13] However, in our study, the activity of GOD reached up to 107.18 U/mL when expressing GOD in the P. pastoris SMD1168 strain with the promoter pGAP under the culture condition of 30 °C and pH 6. This result indicates that the constructed P. pastoris SMD1168-GOD strain has valuable application to express A. niger GOD.
Conclusions
A recombinant plasmid pGAPZαA-GOD, which contained the GOD gene from A. niger accc30161 strain and pGAP promoter, was successfully constructed and was then successfully transformed to P. pastoris SMD1168 to obtain the P. pastoris SMD1168-GOD strain. The recombinant GOD protein was highly expressed in the constructed P. pastoris SMD1168-GOD strain and had high enzyme activity. To the best of our knowledge, this is the first report to construct the P. pastoris SMD1168-GOD strain for high expression of A. niger GOD. Our findings may provide experimental evidence for the large-scale expression and production of recombinant GOD protein.
All authors declare no non-financial competing interests.
Disclosure statement
All authors declare no financial competing interests.
Additional information
Funding
References
- Zeeb B, Fischer L, Weiss J. Stabilization of food dispersions by enzymes. Food Funct. 2014;5(2):198–213.
- Ang LF, Por LY, Yam MF. Development of an amperometric-based glucose biosensor to measure the glucose content of fruit. PLoS One [Internet]. 2015 [ cited 2015 Nov 30];10(3):e0111859. Available from: http://dx.doi.org/10.1371/journal.pone.0111859.
- Ayenimo JG, Adeloju SB. Inhibitive potentiometric detection of trace metals with ultrathin polypyrrole glucose oxidase biosensor. Talanta. 2015;137:62–70.
- Wong CM, Wong KH, Chen XD. Glucose oxidase: natural occurrence, function, properties and industrial applications. Appl Microbiol Biotechnol. 2008;78(6):927–938.
- Canli O, Kurbanoglu EB. Utilization of ram horn peptone in the production of glucose oxidase by a local isolate Aspergillus niger OC-3. Prep Biochem Biotechnol. 2011;41(1):73–83.
- V Garay-Flores R, P Segura-Ceniceros E, De León-Gámez R, et al. Production of glucose oxidase and catalase by Aspergillus niger free and immobilized in alginate-polyvinyl alcohol beads. J Gen Appl Microbiol. 2014;60(6):262–269.
- Gasser B, Prielhofer R, Marx H, et al. Pichia pastoris: protein production host and model organism for biomedical research. Future Microbiol. 2013;8(2):191–208.
- Weinacker D, Rabert C, Zepeda AB, et al. Applications of recombinant Pichia pastoris in the healthcare industry. Braz J Microbiol. 2014;44(4):1043–1048.
- Byrne B. Pichia pastoris as an expression host for membrane protein structural biology. Curr Opin Struct Biol. 2015;32C:9–17.
- Guo Y, Lu F, Zhao H, et al. Cloning and heterologous expression of glucose oxidase gene from Aspergillus niger Z-25 in Pichia pastoris. Appl Biochem Biotechnol. 2010;162(2):498–509.
- Mattanovich D, Branduardi P, Dato L, et al. Recombinant protein production in yeasts. Methods Mol Biol. 2012;824(17):329–358.
- Kovačević G, Blažić M, Draganić B, et al. Cloning, heterologous expression, purification and characterization of M12 mutant of Aspergillus niger glucose oxidase in yeast Pichia pastoris KM71H. Mol Biotechnol. 2014;56(4):305–311.
- Meng Y, Zhao M, Yang M, et al. Production and characterization of recombinant glucose oxidase from Aspergillus niger expressed in Pichia pastoris. Lett Appl Microbiol. 2014;58(4):393–400.
- Gietz RD, Woods RA. Genetic transformation of yeast. Biotechniques. 2001;30(4):816–828.
- Frederick KR, Tung J, Emerick RS, et al. Glucose oxidase from Aspergillus niger. Cloning, gene sequence, secretion from Saccharomyces cerevisiae and kinetic analysis of a yeast-derived enzyme. J Biol Chem. 1990;265(7):3793–3802.
- Whittington H, Kerry-Williams S, Bidgood K, et al. Expression of the Aspergillus niger glucose oxidase gene in A. niger, A. nidulans and Saccharomyces cerevisiae. Curr Genet. 1990;18(6):531–536.
- Kelle S, Zelena K, Krings U, et al. Expression of soluble recombinant lipoxygenase from Pleurotus sapidus in Pichia pastoris. Protein Expr Purif. 2014;95:233–239.
- Zhou X, Yu Y, Tao J, et al. Production of LYZL6, a novel human c-type lysozyme, in recombinant Pichia pastoris employing high cell density fed-batch fermentation. J Biosci Bioeng. 2014;118(4):420–425.
- Wang S, Li P, Su J, et al. Characterization and expression of glucosamine-6-phosphate synthase from Saccharomyces cerevisiae in Pichia pastoris. Biotechnol Lett. 2014;36(10):2023–2028.
- Assiri AS, El-Gamal BA, Hafez EE, et al. Production of recombinant streptokinase from Streptococcus pyogenes isolate and its potential for thrombolytic therapy. Saudi Med J. 2014;35(12):1482–1488.
- Daly R, Hearn MT. Expression of heterologous proteins in Pichia pastoris: a useful experimental tool in protein engineering and production. J Mol Recognit. 2005;18(2):119–138.
- Vogl T, Glieder A. Regulation of Pichia pastoris promoters and its consequences for protein production. New Biotechnol. 2013;30(4):385–404.
- Vogl T, Hartner FS, Glieder A. New opportunities by synthetic biology for biopharmaceutical production in Pichia pastoris. Curr Opin Biotechnol. 2013;24(6):1094–1101.
- Wang X, Sun Y, Ke F, et al. Constitutive expression of Yarrowia lipolytica lipase LIP2 in Pichia pastoris using GAP as promoter. Appl Biochem Biotechnol. 2012;166(5):1355–1367.
- Zhang AL, Luo JX, Zhang TY. Recent advances on the GAP promoter derived expression system of Pichia pastoris. Mol Biol Rep. 2009;36(6):1611–1619.
- Zhang AL, Zhang TY, Luo JX, et al. Constitutive expression of human angiostatin in Pichia pastoris by high-density cell culture. J Ind Microbiol Biotechnol. 2007;34(2):117–122.
- Yang CH, Huang YC, Chen CY, et al. Expression of Thermobifida fusca thermostable raw starch digesting alphal-amylase in Pichia pastoris and its application in raw sago starch hydrolysis. J Ind Microbiol Biotechnol. 2010;37(4):401–406.
- Qiao J, Rao Z, Dong B, et al. Expression of Bacillus subtilis MA139 beta-mannanase in Pichia pastoris and the enzyme characterization. Appl Biochem Biotechnol. 2010;160(5):1362–1370.
- Zhang ZH, Yang HS, Zhang AL. Using pGAP promoter to express of laccase gene from Bacillus subtilis in P. pastoris. Biotechnology. 2011;21(6):24–27.
- Liu ZW, Yin HX, Yi XP, et al. Constitutive expression of barley α-amylase in Pichia pastoris by high-density cell culture. Mol Biol Rep. 2012;39(5):5805–5810.
- Nie Y, Huang M, Lu J, et al. Impacts of high β-galactosidase expression on central metabolism of recombinant Pichia pastoris GS115 using glucose as sole carbon source via (13)C metabolic flux analysis. J Biotechnol. 2014;187:124–134.
- Yamaguchi M, Tahara Y, Nakano A, et al. Secretory and continuous expression of Aspergillus niger glucose oxidase gene in Pichia pastoris. Protein Expr Purif. 2007;55(2):273–278.