
ABSTRACT
Introducing various mutations at multiple specific sites within a gene requires multiple steps of DNA manipulation, which is the initial, but limiting step of protein structure–function studies. In the present work, we standardized a simple and fast procedure to perform site-directed mutagenesis, multiple-site fragment deletion, insertion and substitution mutagenesis by a modified version of overlap extension polymerase chain reaction (PCR). In this procedure, target genes divided into several fragments based on the site of mutagenesis are amplified and annealed with their complementary overhanging, followed by extension and amplification to full-length gene with expected mutation(s) by PCR. Vectors inserted with the modified target gene are screened by colony PCR. By using the standardized procedure, we have easily generated single-site mutations, replaced/deleted DNA fragment into/from a target gene and engineered a cysteine-free protein. Practically, the standardized procedure provides an efficient choice for almost all kinds of mutagenesis, especially for multiple-site and large DNA fragment modification mutagenesis. Therefore, this method can be utilized to analyze protein structure and function, to optimize codons of genes for protein expression and to assemble genes of interest.
| Abbreviations | ||
| PCR: | = | polymerase chain reaction; |
| OE-PCR: | = | overlap extension by polymerase chain reaction; |
| ERCC8: | = | Excision Repair Cross-Complementation Group 8; |
| RE: | = | DNA fragment for replacement mutagenesis; |
| IN: | = | DNA fragment for insertion mutagenesis; |
| DE: | = | DNA fragment for deletion mutagenesis; |
| bp: | = | base pair; |
| kbp: | = | kilobase pair |
Introduction
Site-specific mutagenesis of DNA, which allows deleting, inserting or substituting multiple-site DNA fragments, is an important tool in molecular biology, genetic engineering, biochemistry and protein engineering. Among other site-directed mutagenesis strategies [Citation1–8], the QuikChange™ Site-Directed Mutagenesis System developed by Stratagene (La Jolla, CA, USA) has been widely used and considered as a powerful tool for site-directed mutagenesis, which works via a pair of complementary primers containing the wanted mutation. However, the presence of parent template has shown to give high false positives. Primer dimers and tandem repeats of primers appear with a high frequency in some cases and therefore affect the generation of expected mutations [Citation9]. In addition, DNA sequencing is required to confirm the mutants. As the originally developed QuikChange™ cannot introduce multiple mutations as well as long DNA fragment deletion, insertion or replacement mutations, a modified version of the kit (QuikChange™ Multi Site-Directed Mutagenesis kit) has been released and some other adaptations have been reported afterwards [Citation10–19]. However, these procedures need special care in the design of primers under some strict rules and/or can only be used for certain specific kinds of mutagenesis. So far, there still lacks a simple and highly efficient method that allows deleting, inserting or substituting multiple-site DNA fragments. These conundrums prompted us to develop a new method of mutagenesis that would follow the rule of simplicity but have promising efficiency and applicability.
Overlap extension by polymerase chain reaction (OE-PCR), described by [Citation20], has been developed as a powerful tool to generate mutations [Citation21–26]. With this technique, mutated DNA fragments are generated and ligated through the overlapping ends, in which the overlapping ends anneal, allowing the 3′ overlap of each strand to serve as the primer for the extension of the complementary strand [Citation20,Citation27]. In this work, we described a modified OE-PCR method for multiple-site fragment deletion, insertion and substitution mutagenesis. By this work, we intended to standardize a fast and simple procedure with the aim of achieving a high mutagenic efficiency for any kind of expected mutagenesis listed above. The present work shows how our standardized procedure was successfully used to achieve various mutations, including single-site mutations, multiple-site mutations and insertion/deletions in a human gene ERCC8 (Excision Repair Cross-Complementation Group 8).
Materials and methods
Materials
Human cDNAs was purchased from Takara Bio (Madison, WI, USA). KOD hot start DNA polymerase was purchased from Novagen (Billerica, MA, USA), restriction endonucleases, DNA marker, Taq DNA polymerase and T4 DNA ligase from New England Biolabs (Ipswich, MA, USA) and cloning kits from Qiagen (Germantown, MD, USA). Vector pET30a, host strain Escherichia coli DH5α was obtained from Invitrogen Corp. (Waltham, MA, USA). Oligonucleotide primers were purchased from Invitrogen Corp. The PCR purification kit and gel extraction kit were purchased from Qiagen. The plasmids were isolated using a QIAprep Spin Miniprep Kit (Qiagen). Samples with mutations were sent to Shanghai Sunny Biotechnology Co. Ltd. (Shanghai, China) for sequencing.
Vector
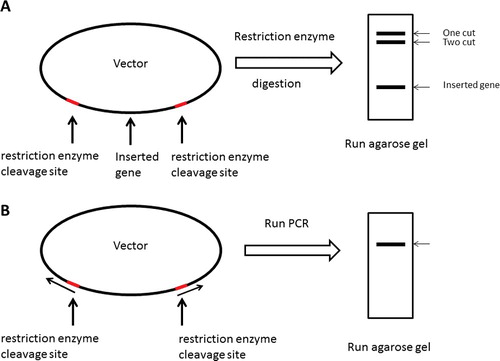
In order to generate a parent vector necessary for annealing of the mutated gene of interest, either of two methods was used. As shown in (A), the vector was gel-purified upon restriction digestion and removal of an already existing insertion but with restriction sites intact. PCR was used as the second approach to generate a linear vector with two restriction recognition sites at each end ((B)). After purification by 1% agarose gel electrophoresis, PCR products were digested by restriction enzymes. The purified product was then digested by Dpn1 and allowed to go through another purification step using a QIAquick® PCR purification kit to eliminate the residual contaminations of the template.
Figure 1. Schematic presentations of parent vector generation processes. The parent vector was generated by restriction digestion of a vector that already harbours an inserted gene (A), or by PCR method (B). The grey line shows restriction enzyme cleavage site. For details, see text.
Touchdown PCR
PCR reactions were performed to introduce mutations at the specific points in a final volume of 50 μL using MyCycler™ thermocycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA), KOD hot start DNA polymerase (Novagen), and the primer pair as shown in . After the initial denaturation step at 98 °C for 5 min, the PCR was conducted for 20 cycles with denaturation at 98°C for 20 s, primer annealing from 60 to 50 °C with a step of −0.5 °C each cycle for 20 °C and extension at 72 °C using a time of 1 min/kbp, following by 10 cycles with a fixed anneal temperature at 52 °C. When all cycles were completed, the samples were kept at 72 °C for 10 min to finish all of DNA synthesis as indicated in .
Table 1. Primers used for mutagenesis.
Table 2. Thermocycling conditions for touchdown PCR.
Colony PCR
For each transformation, we selected at least eight colonies at random and performed colony PCR for determining insertion, with five units of Taq DNA polymerase (NEB) and 1×ThermoPol® Buffer (NEB) in the presence of 200 μmol/L of deoxynucleoside triphosphates (dNTP), 1 mmol of a primer from the vector and a primer from the insert gene and a small amount of cells picked from the colony in a final volume of 20 μL. The colony PCR reaction programs were optimized as follows: 95 °C for 2 min, then 25 cycles of 95 °C for 30 s for denaturation, 50 °C for 30 s and 68 °C using a time of 1 min/kb, followed by 68 °C for 10 min for the final extension as shown in .
Table 3. Thermocycling conditions for colony PCR.
Results and discussion
Single-site mutagenesis
In this study, we used the ERCC8 gene (NCBI Reference Sequence: NM_000082.3, 1191 bp) which encodes DNA excision repair protein ERCC-8 in humans (NCBI Reference Sequence: NP_000073.1, 396 aa) as an example (see Supplementary Figure S1). The ERCC8 gene was amplified from a human cDNA library (Takara Bio) and inserted into pET30a between EcoRI and HindIII restriction recognition sites.
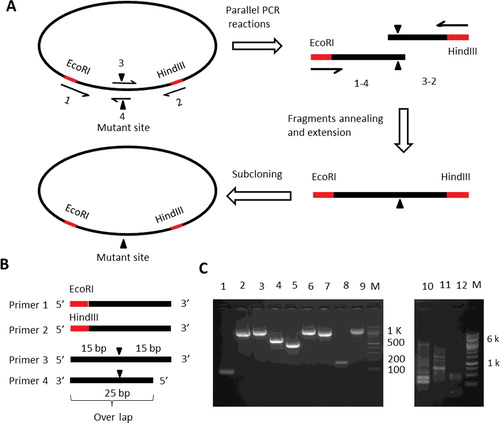
shows the scheme of the PCR amplification processes (A) and the primers design (B) used to generate single-site mutation. To generate a single-site mutation ((A), filled black triangle), we designed four primers: primer 1, forward primer, which contains a ∼25 bp homologous sequence to the positive strand and an additional restriction enzyme cut site at the 5′ end; primer 2, reverse primer, which contains a ∼25 bp sequence that is homologous to the negative strand and an additional restriction enzyme cut site at the 3′ end; primer 3, a ∼30 bp homologous sequence to the positive strand with mutated bases in the primer centre; and primer 4, a ∼25 bp complementary sequence to primer 3 at the 5′ end ((B)). First, we used two pairs of primers, primer 1/primer 4 and primer 3/primer 2 to generate two DNA fragments using the target gene as the template. The PCR products were gel-purified. These two DNA fragments with cohesive 5′ or 3′ end were therefore able to anneal together with their complementary overhanging cohesive ends, which were then extended and amplified by PCR using the primer pair 1/2 to get a full-length gene with the expected mutation ((A)).
Figure 2. Schematic presentations of single-site mutagenesis PCR amplification processes. (A) The scheme of the method used to generate single-site mutations. (B) Schematic diagram of the primer design for site-directed mutagenesis. Black filled triangles show the location of mutations. Grey filled rectangles show the restriction digestion sites. (C) Agarose gel electrophoresis of the PCR reactions for single-site mutagenesis. The primers used for DNA synthesis and the expected length of the DNA products are: Lane 1, ERCC8EcoRIfw/ERCC8S23Crv, 84 bp; Lane 2, ERCC8S23Cfw/ERCC8HindIIIrv, 1149 bp; Lane 3, ERCC8EcoRIfw/ERCC8HindIIIrv, 1209 bp; Lane 4, ERCC8EcoRIfw/ERCC8L202Crv, 652 bp; Lane 5, ERCC8L202Cfw/ERCC8HindIIIrv, 582 bp; Lane 6, ERCC8EcoRIfw/ERCC8HindIIIrv, 1209 bp; Lane 7, ERCC8EcoRIfw/ERCC8Y350Crv, 1064 bp; Lane 8, ERCC8Y350Cfw/ERCC8HindIIIrv, 170 bp; Lane 9, ERCC8EcoRIfw/ERCC8HindIIIrv, 1209 bp; Lane 10, S23Cfw/S23Crv, ∼6.6 kbp; Lane 11, K212Cfw/K212Crv, ∼6.6 kbp; Lane 12, Y350Cfw/Y350Crv, ∼6.6 kbp. M, DNA ladder (Qiagen, Germantown, MD, USA). DNA samples were electrophoresed in 2% agarose gel for short fragments (left panel) or 1% agarose gel for longer fragments (right panel).
In our case, we used EcoRI and HindIII to digest and ligate the target gene into the recipient plasmid. We designed primer 1 (ERCC8EcoRIfw), forward primer, which used the sequence 5'-ATGCTGGGGTTTTTGTCCGCAC-3' for the region that binds the open reading frame (ORF), and we then added the EcoRI restriction site (GAATTC) plus three bases (CCG) flanking that site to the 5′ end of this primer [Citation28], making our forward primer 5′-CCGGAATTCATGTGGCATATCTCGAAGTAC-3′. We also designed primer 2 (ERCC8HindIIIrv), reserve primer, which used the sequence 5′-TCATCCTTCTTCATCACTGCTGC-3′ for the region that binds the ORF, and we then added the HindIII restriction site (AAGCTT) plus three bases (CCC) flanking that site to the 5′ end of this primer, making our reserve primer 5′-CCCAAGCTTTCATCCTTCTTCATCACTGCTGC-3′. To help the mutagenic oligonucleotide primers design, we used an automated web site (http://bioinformatics.org/primerx). In this method, the melting temperature, the GC content, the length and the complementarity of primers are not severely limited. In general, the mutation site can be placed as close as 10 bases away from the 5′-terminus or 3′-terminus, and at least one G or C should be placed at the end of each terminus. To facilitate the primer design, we always designed two mutagenic oligonucleotide primers: one containing the mutation in the middle of the primer with ∼10–15 bases of correct sequence on both sides and at least one G or C at the end of each terminus; and the other one containing ∼25 bp primer–primer complementary (overlapping) sequences at the 5′ end. The schematic presentation of our primer design is shown in (B). To evaluate the efficiency of this method for the generation of mutation, three residues at different positions of the ERCC-8 protein, i.e. S23, K212 and Y350, were selected for cysteine-replacement mutagenesis. The properties of the designed primers are shown in and Supplementary Figure S2.
Two-step PCR reactions were performed to introduce a mutation at a specific point using the primer pairs of ERCC8EcoRIfw/ERCC8S23Crv, ERCC8S23Cfw/ERCC8HindIIIrv, ERCC8EcoRIfw/ERCC8K212Crv, ERCC8K212Cfw/ERCC8HindIIIrv, ERCC8EcoRIfw/ERCC8Y350Crv and ERCC8Y350Cfw/ERCC8HindIIIrv (). After the PCR, the DNA products were gel-purified. The two PCR products with cohesive ends were therefore able to anneal together with their complementary overhanging, cohesive ends, which were then extended and amplified by PCR as indicated in using the primer pair ERCC8EcoRIfw/ERCC8HindIIIrv for a full-length gene with the desired mutation ((C)). The DNA products were gel-purified and digested by EcoRI/HindIII restriction enzymes (NEB). The genes containing the desired mutations were ligated into a backbone vector, and then transformed to DH5α competent cells. Colony PCR was used for determining insertion. The plasmids were isolated using a QIAprep Spin Miniprep Kit (Qiagen). Mutations were further confirmed by DNA sequencing.
In comparison, completely overlapping primers designed as recommended in the QuikChange™ manual as described in [Citation29] were also tested in the same positions as described before (S23C, K212C and Y350C) using primer pairs S23Cfw/S23Crv, K212Cfw/K212Crv and Y350Cfw/Y350Crv (). All of the reactions failed to produce any amplification product ((C), right panel), even though these primers were designed according to the protocols of the standard QuikChange™ mutagenesis protocol. For our method, the primers are designed according to the rules for QuikChange™ and therefore no special care for primers design is required. As only short DNA fragments were amplified, the PCR amplification using these primers showed high efficiency. There is no need of plasmid as the parental template, which eliminates the potential for the recovery of the parental DNA. To reduce the probability of the contamination from the backbone vector resulting from incomplete digestion, the plasmid inserted with the target gene was digested with restriction enzymes, followed by gel-purification. In this way, it would be much easier to separate the vectors that have been digested, from those vectors that have not been completely digested.
Multiple-site mutagenesis
shows the flow chart of the generation of multiple-site mutations. The ERCC-8 protein has 13 cysteines, i.e. C84, C88, C157, C171, C178, C222, C252, C288, C301, C303, C339, C340 and C356 (). In this study, the 13 cysteines were replaced by serine to get a cysteine-free protein. As the codons coding for C84 and C88, C157, C171 and C178, C288, C301 and C303, and C339, C340 and C356 residues are too close to amplify by regular PCR, they can be grouped together. The whole gene can be divided into seven fragments by six cysteine-encoding groups, i.e. C84/C88, C157/C171/C178, C222, C252, C288/C301/C303 and C339/C340/C356 (). The primer pairs used to mutate cysteine to serine were designed according to the standard QuikChange™ Mutagenesis protocol (). Seven parallel PCR reactions were performed to amplify each DNA fragment by using the primers as shown in and . The amplified products were separated by 1% agarose gel electrophoresis and purified by gel extraction. DNA fragments ① and ② were annealed together with their complementary overhanging, cohesive ends, and extended and amplified by PCR using the primer pairs as shown in and to generate DNA ⑧ following purification by 1% agarose electrophoresis gel and DNA extraction. By using the same procedure, DNA fragments ⑨ and ⑩ can be obtained (). Next, DNA fragments ⑧ and ⑨ were annealed and extended to generate DNA fragment ⑪, and DNA fragments ⑩ and ⑦ were used to generate DNA fragment ⑫. Finally, DNA fragment ⑪ and DNA fragment ⑫ were used to engineer a full-length gene with the desired multiple-site mutations. Agarose gel electrophoresis of the synthesized DNA fragments, with the primer pairs used to synthesize the desired DNA fragments, and the expected length of the DNA products are shown in . The gene with multiple-site mutations was digested by EcoRI/HindIII restriction enzymes (NEB), was subcloned into a backbone vector, which was digested by the same restriction enzymes, and then transformed to DH5α competent cells. The presence or absence of insert DNA in the plasmid constructs was determined by colony PCR (see ).
Figure 3. Flow chart of the multiple-site mutagenesis method. Black filled triangles show the location of mutations. Grey filled rectangles show the restriction digestion sites. The grey lines with arrowhead show the primers used for DNA synthesis. The time required for each step is shown on the right.
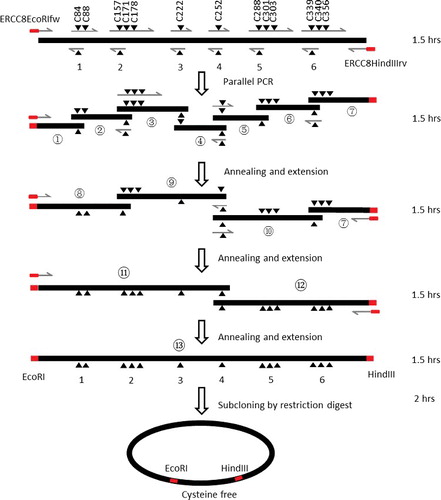
Figure 4. Agarose gel electrophoresis of the PCR reactions for multiple-site mutagenesis following the procedure described in . The primers used for DNA synthesis and the expected length of the DNA products are: Lane 1, ERCC8EEcoRIfw/ERCC8EC84SC88Srv, 270 bp; Lane 2, ERCC8EC84SC88Sfw/ERCC8EC157SC171SC178Srv, 244 bp; Lane 3, ERCC8EC157SC171SC178Sfw/ERCC8C222Srv, 220 bp; Lane 4, ERCC8C222Sfw/ ERCC8C252Srv, 116 bp; Lane 5, ERCC8C252Cfw/ERCC8C288SC301SC303Srv, 134 bp; Lane 6, ERCC8C288SC301SC303Sfw/ERCC8C339SC340SC356Srv, 181 bp; Lane 7, ERCC8C339SC340SC356Sfw/ERCC8HindIIIrv, 204 bp; Lane 8, ERCC8EEcoRIfw/ERCC8EC157SC171SC178Srv, 587 bp; Lane 9, ERCC8EC157SC171SC178Sfw/ERCC8C252Srv, 310 bp; Lane 10, ERCC8C252Sfw/ERCC8C339SC340SC356Srv, 289 bp; Lane 11, ERCC8EEcoRIfw/ERCC8C252Srv, 773 bp; Lane 12, ERCC8C252Sfw/ERCC8HindIIIrv, 463 bp; Lane 13, ERCC8EEcoRIfw/ERCC8HindIIIrv, 1209 bp. M, DNA ladder (Qiagen, Germantown, MD, USA). DNA samples were electrophoresed in 2% agarose gel.

In comparison, for the QuickChange™ site-directed mutagenesis method, the cysteine residues have to be mutated one by one and it may need at least 13 days to get the same mutant. Moreover, DNA sequencing is required for each mutation step to confirm the positive mutation. It normally takes much longer time to get multiple-site mutagenesis by using the QuickChange™ site-directed mutagenesis method. Significantly, multiple-site mutagenesis can be achieved in a quite short time with our described method (). The advantage of this method over other methods is its simplicity and saving of time, since DNA sequencing is not required at each step.
Replacement, insertion and deletion mutagenesis
shows the scheme that can be used to generate replacement, insertion or deletion mutations. In this study, we planned to replace a 200-bp DNA fragment from the ERCC8 gene by another DNA fragment, or insert a 200-bp DNA fragment into the ERCC8 gene, or remove a 200-bp DNA fragment from the ERCC8 gene (Figure S3).
Figure 5. Schematic presentations of deletion, replacement and insertion mutagenesis PCR amplification processes. Black filled triangles show the location of mutations. Grey filled rectangles show the restriction digestion sites. The lines with arrowhead show the primers used for DNA synthesis. RE indicates the DNA fragment that is used to replace another DNA fragment. IN indicates the DNA fragment that will be inserted into another gene. DE indicates the DNA fragment that will be deleted.
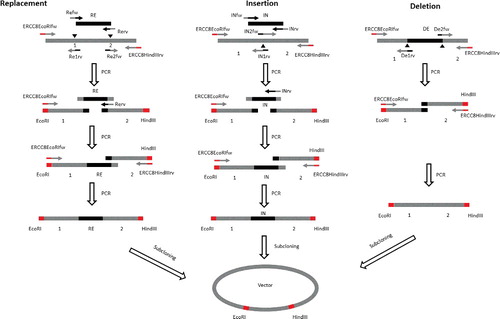
As shown in , to replace a 200-bp DNA fragment (between 1 and 2, Figure S3) from the ERCC8 gene with another one (RE), three parallel PCR reactions were performed to amplify each DNA fragment by using the primer pairs ERCC8EcoRIfw/Re1rv, Refw/Rerv and Re2fw/ERCC8HindIIIrv () to generate DNA fragment ‘1’, which has an EcoRI recognition site at the 5′ end and a complementary overhanging to the RE DNA fragment at the 3′ end; fragment ‘2’, which has a HindIII recognition site at the 3′ end and a complementary overhanging to the RE DNA fragment at the 5′ end; and fragment ‘RE’, which has a complementary overhanging to DNA fragment ‘1’ at the 5′ end and a complementary overhanging to DNA fragment ‘2’ at the 3′ end. Amplified products were separated by 1% agarose gel electrophoresis and purified by gel extraction. DNA fragments ‘1’ and ‘RE’ were annealed together with their complementary overhanging, cohesive ends, and extended and amplified by PCR using the primer pair EcoRIfw/Rerv to generate DNA fragment ‘1+RE’, which has an EcoRI recognition site at the 5′ end and a complementary overhanging to DNA fragment ‘2’ at the 3′ end. DNA fragment ‘1+RE’ was then purified by 1% agarose electrophoresis gel and DNA extraction. Next, DNA fragments ‘1+RE’ and ‘2’ were annealed together with their complementary overhanging, cohesive ends, and extended and amplified by PCR using the primer pair ERCC8EcoRIfw/ERCC8HindIIIrv to generate DNA fragment ‘1+RE+2’, which has an EcoRI recognition site at the 5′ end, a HindIII recognition site at the 3′ end and the DNA sequence between ‘1’ and ‘2’ replaced by the RE DNA fragment. The amplified products were separated by 1% agarose gel electrophoresis and purified by gel extraction. Agarose gel electrophoresis of the amplified DNA is shown in .
Figure 6. Agarose gel electrophoresis of the PCR reactions for deletion, replacement, and insertion mutagenesis following the processes as described in . The primers used for DNA synthesis and the expected length of the DNA products are: Lane 1, EcoRIfw/Re1rv, 309 bp; Lane 2, Refw/Rerv, 248 bp; Lane 3, Re2fw/HindIIIrv, 724 bp; Lane 4, EcoRIfw/Rerv, 533 bp; Lane 5, EcoRIfw/HindIIIrv, 1209 bp; Lane 6, EcoRIfw/IN1rv, 629 bp; Lane 7, IN2fw/HindIIIrv, 628 bp; Lane 8, INfw/Inrv, 248 bp; Lane 9, EcoRIfw/Inrv, 829 bp; Lane 10, EcoRIfw/HindIIIrv, 1409 bp; Lane 11, EcoRIfw/De1rv, 310 bp; Lane 12, De2fw/HindIIIrv, 732 bp; Lane 13, EcoRIfw/HindIIIrv, 1009 bp. M, DNA ladder. DNA samples were electrophoresed in 2% agarose gel.
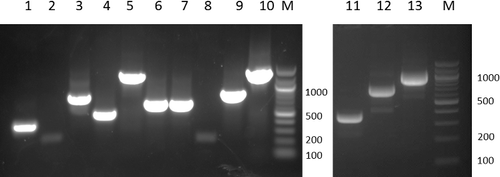
To insert a 200-bp DNA fragment (IN) into the ERCC8 gene, three parallel PCR reactions were performed to amplify each DNA fragment by using the primer pairs, ERCC8EcoRIfw/IN1rv, INfw/INrv and IN2fw/ERCC8HindIIIrv (, and Supplementary Figure S3), to generate DNA fragment ‘1’, which has an EcoRI recognition site at the 5′ end and a complementary overhanging to the IN DNA fragment at the 3′ end; ‘2’, which has a HindIII recognition site at the 3′ end and a complementary overhanging to the IN DNA fragment at the 5′ end; and ‘IN’, which has a complementary overhanging to DNA fragment ‘1’ at the 5′ end and a complementary overhanging to DNA fragment ‘2’ at the 3’ end. The amplified products were separated by 1% agarose gel electrophoresis and purified by gel extraction. DNA fragments ‘1’ and ‘IN’ were annealed together with their complementary overhanging, cohesive ends, and extended and amplified by following PCR using the primer pair ERCC8EcoRIfw/INrv to generate DNA fragment ‘1+IN’, which has an EcoRI recognition site at the 5′ end and a complementary overhanging to the ‘2’ DNA fragment at the 3′ end. DNA fragment ‘1+IN’ was then purified by 1% agarose gel electrophoresis and DNA extraction. Next, DNA fragments ‘1+IN’ and ‘2’ were annealed together with their complementary overhanging, cohesive ends, and extended and amplified by PCR using the primer pair ERCC8EcoRIfw/ERCC8HindIIIrv to generate DNA fragment ‘1+IN+2’, which has an EcoRI recognition site at the 5′ end, a HindIII recognition site at the 3′ end, and the IN DNA fragment inserted into the target gene. The amplified products were separated by 1% agarose gel electrophoresis and purified by gel extraction. Agarose gel electrophoresis of the amplified DNA is shown in .
To delete a 200-bp DNA fragment (DE) from the ERCC8 gene between ‘1’ and ‘2’ (, Supplementary Figure S3), two parallel PCR reactions were performed to amplify each DNA fragment by using the primer pairs ERCC8EcoRIfw/De1rv and De2fw/ERCC8HindIIIrv to generate DNA fragment ‘1’, which has an EcoRI recognition site at the 5′ end and a complementary overhanging to ‘2’ DNA fragment at the 3′ end, ‘2’, which has a HindIII recognition site at the 3′ end and a complementary overhanging to ‘1’ DNA fragment at the 5′ end. The amplified products were separated by 1% agarose gel electrophoresis and purified by gel extraction. DNA fragments ‘1’ and ‘2’ were annealed together with their complementary overhanging, cohesive ends, and extended and amplified by following PCR using the primer pair ERCC8EcoRIfw/ERCC8HindIIIrv to generate a modified gene, which has an EcoRI recognition site at the 5′ end, a HindIII recognition site at the 3′ end and a DNA fragment deletion in the target location. The amplified products were separated by 1% agarose gel electrophoresis and purified by gel extraction. Agarose gel electrophoresis of the amplified DNA is shown in .
Finally, the genes with multiple-site mutations, which were digested by EcoRI/HindIII restriction enzymes (NEB), were ligated into a backbone vector, which was digested by the same restriction enzymes using T4 ligase, and then transformed to DH5α competent cells. The presence or absence of inserted DNA in the plasmid constructs was determined by colony PCR.
In the present work, we successfully substituted, inserted and deleted a 200-bp DNA fragment to or from the target gene ( and ). As we never use a complementary primer pair to amplify DNA, this design would eliminate the problems of primer pair self-annealing that are always associated with the complementary primer pair design. For the present method, the Tm values are not strictly limited. A DNA fragment at any position of the target gene can be deleted or replaced without limitation. The length of the DNA fragments for replacement, deletion or insertion is also not strictly limited.
Conclusions
As a result of the present work, we have standardized a procedure for rapid and efficient multiple-site fragment deletion, insertion and substitution mutagenesis based on modified overlap extension PCR. This method utilizes a new DNA fragment-designing scheme, which facilitated the design of primers and the PCR procedure, and enhanced the overall efficiency and reliability. By using this method, we successfully generated single/multiple-site mutations, deletion, insertion and substitution mutations. The results demonstrated that this protocol would not increase any reagent costs but increased the overall positive rates. It provided an efficient choice, especially for multiple-site, or large DNA fragment modification mutagenesis of DNAs.
Supplementary_Data.pdf
Download PDF (309.7 KB)Acknowledgments
We would like to thank Dr E. Padros (Department of Biochemistry and Molecular Biology, Medical School, and Center of Biophysics, Autonomous University of Barcelona) for critically reading of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Higuchi R, Krummel B, Saiki RK. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–7367.
- Warrens AN, Jones MD, Lechler RI. Splicing by overlap extension by PCR using asymmetric amplification: an improved technique for the generation of hybrid proteins of immunological interest. Gene. 1997;186(1):29–35.
- Chiu J, March PE, Lee R, et al. Site-directed, Ligase-Independent Mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 2004;32(21):e174.
- Allemandou F, Nussberger J, Brunner HR, et al. Rapid site-directed mutagenesis using two-PCR-generated DNA fragments reproducing the plasmid template. J Biomed Biotechnol. 2003;2003(3):202–207.
- Tyagi R, Lai R, Duggleby RG. A new approach to ‘megaprimer’ polymerase chain reaction mutagenesis without an intermediate gel purification step. BMC Biotechnol. 2004;4:2.
- Kirsch RD, Joly E. An improved PCR-mutagenesis strategy for two-site mutagenesis or sequence swapping between related genes. Nucleic Acids Res. 1998;26(7):1848–1850.
- Kegler-Ebo DM, Docktor CM, DiMaio D. Codon cassette mutagenesis: a general method to insert or replace individual codons by using universal mutagenic cassettes. Nucleic Acids Res. 1994;22(9):1593–1599.
- Zoller MJ, Smith M. Oligonucleotide-directed mutagenesis using M13-derived vectors: an efficient and general procedure for the production of point mutations in any fragment of DNA. Nucleic Acids Res. 1982;10(20):6487–6500.
- Edelheit O, Hanukoglu A, Hanukoglu I. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure–function studies. BMC Biotechnol. 2009;9(1):61.
- Mikaelian I, Sergeant A. A general and fast method to generate multiple site directed mutations. Nucleic Acids Res. 1992;20(2):376.
- Jensen PH, Weilguny D. Combination primer polymerase chain reaction for multi-site mutagenesis of close proximity sites. J Biomol Tech. 2005;16(4):336–340.
- Young L, Dong Q. TAMS technology for simple and efficient in vitro site-directed mutagenesis and mutant screening. Nucleic Acids Res. 2003;31(3):e11. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC149223/
- Kim YG, Maas S. Multiple site mutagenesis with high targeting efficiency in one cloning step. Biotechniques. 2000;28(2):196–198.
- Wang W, Malcolm BA. Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. Biotechniques. 1999;26(4):680–682.
- Liu H, Naismith JH. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008;8:91. Available from: http://bmcbiotechnol.biomedcentral.com/articles/10.1186/1472-6750-8-91
- Liang X, Peng L, Li K, et al. A method for multi-site-directed mutagenesis based on homologous recombination. Anal Biochem. 2012;427(1):99–101.
- Guo W, Sun S, Xie B, et al. T4 DNA polymerase improves the efficiency of multiple site-directed mutagenesis. Biotechnol Biotechnol Equip. 2016;30(4):721–725.
- Trehan A, Kielbus M, Czapinski J, et al. REPLACR-mutagenesis, a one-step method for site-directed mutagenesis by recombineering. Sci Rep. 2016;6:19121.
- Mitchell LA, Cai Y, Taylor M, et al. Multichange isothermal mutagenesis: a new strategy for multiple site-directed mutations in plasmid DNA. ACS Synth Biol. 2013;2(8):473–477.
- Ho SN, Hunt HD, Horton RM, et al. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77(1):51–59.
- Bryksin AV, Matsumura I. Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. Biotechniques. 2010;48(6):463–465.
- Urban A, Neukirchen S, Jaeger KE. A rapid and efficient method for site-directed mutagenesis using one-step overlap extension PCR. Nucleic Acids Res. 1997;25(11):2227–2228.
- An Y, Ji J, Wu W, et al. A rapid and efficient method for multiple-site mutagenesis with a modified overlap extension PCR. Appl Microbiol Biotechnol. 2005;68(6):774–778.
- Lee J, Shin MK, Ryu DK, et al. Insertion and deletion mutagenesis by overlap extension PCR. Methods Mol Biol. 2010;634:137–146.
- Waneskog M, Bjerling P. Multi-fragment site-directed mutagenic overlap extension polymerase chain reaction as a competitive alternative to the enzymatic assembly method. Anal Biochem. 2014;444:32–37.
- Williams EM, Copp JN, Ackerley DF. Site-saturation mutagenesis by overlap extension PCR. Methods Mol Biol. 2014;1179:83–101.
- Ito W, Ishiguro H, Kurosawa Y. A general method for introducing a series of mutations into cloned DNA using the polymerase chain reaction. Gene. 1991;102(1):67–70.
- Jung V, Pestka SB, Pestka S. Efficient cloning of PCR generated DNA containing terminal restriction endonuclease recognition sites. Nucleic Acids Res. 1990;18(20):6156.
- Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004;32(14):e115.