
ABSTRACT
NdmB and ndmD genes encoding N-demethylase (Ndm) B and NdmD from Pseudomonas putida CBB5 were successfully cloned in pET32a, designated as pET32a-ndmB-His-His-ndmD (pET32a-BHHD) and electroporated into Escherichia coli strain BL21 (DE3). Polymerase chain reaction (PCR), restriction enzyme digestion and DNA sequencing were subsequently employed to confirm the success of the procedure. Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) showed that the estimated molecular weight of NdmB and NdmD co-expressed in recombinant E. coli was 35 and 60 kDa, respectively. A one-step purification of Ndms using a Ni-affinity column resulted in a 10.3-fold purification with 33.6% yield. The enzyme activity of NdmB and NdmD showed that 1000 μmol/L theobromine was completely converted into 7-methylxanthine within 90 min by resting cells containing pBHHD.
Introduction
1,3,7-Trimethylxanthine and 3,7-trimethylxanthine (TB) are well known as caffeine and theobromine, respectively. Both of them as well as related purine alkaloids are consumed extensively in the form of coffee, tea, cola, cakes, chocolates and some pharmaceutical products [Citation1,Citation2]. N-demethylation is the principal pathway to metabolize TB and caffeine in many plants and microorganisms [Citation3,Citation4]. 1A2 as well as 2E1, types of hepatic cytochrome P450s, are in charge of this process in humans [Citation5]. Previous research has shown that many microorganisms possess a so-called N-demethylase (Ndms), which catalyzes the N-demethylation process to degrade caffeine as well as methylxanthines [Citation6,Citation7]. Besides, C8-oxidation catalyzed by caffeine oxidases and caffeine dehydrogenase is a second choice in these microorganisms [Citation8–10] ((A)). Yu et al. [Citation9] isolated a unique caffeine-degrading bacterium, Pseudomonas putida CBB5. By successive N-demethylation, the bacterium builds the intact pathway to degrade caffeine and other related methylxanthines [Citation11,Citation12]. Summers et al. [Citation13] have identified and characterized successively Ndm A, B, C, D and E. A similar caffeine metabolic pathway has also been characterized genetically in Pseudomonas sp. CES [Citation14].
Figure 1. Caffeine degradation pathways: (A) C-8 oxidation pathway for degradation of caffeine by Pseudomonas sp. CBB1; (B) N-demethylation pathway for conversion of caffeine into xanthine by Pseudomonas putida CBB5.
Methylxanthines have important pharmacological properties, such as stimulatory effects on the central nervous system, gastrointestinal, cardiovascular, renal and respiratory systems. Therefore, they are commonly used as therapeutic agents [Citation15]. For example, TB is a cough suppressant which can reduce asthma symptoms. 7-Methylxanthine (7-MX) is an efficient agent in retarding axial elongation and myopia progression among myopic children [Citation16]. However, most methylxanthines are difficult to synthesize chemically because selective alkylation of each nitrogen atom is difficult to achieve [Citation4,Citation14], whereas degrading caffeine or TB by microbial or enzyme technology has become a hot topic of research, considering its high-specificity, eco-friendliness and cost-effectiveness. Components such as TB, theophylline (TP) and 7-MX, which are wildly used in the industry and clinic, are produced by this biocatalytic method [Citation17–19].
The recently discovered genes of ndms A, B, C, D and E from P. putida CBB5 provide an excellent strategy for production of specific high-value paraxanthine and 1-, 3- and 7-MX from any of the economic feedstocks including caffeine, TB or TP by precisely engineered Escherichia coli with various gene combinations. For example, the ndmA and ndmD genes were cloned into E. coli, resulting in a bacterial strain that was able to effectively convert caffeine to TB [Citation20] or TP to 3-MX [Citation21]. These several strains mainly catalyzed N1-demethylation reactions, whereas N3-demethylation reactions catalyzed by engineered strains have rarely been reported. Among the Ndms, NdmB catalyzes the specific N3-demethylation of caffeine, 3,7-dimethylxanthine and 3-methylxanthine into 1,7-dimethyl xanthine, 7-methylxanthine and xanthine, respectively, while NdmD couples with NdmB by transferring electrons from nicotinamide adenine dinucleotide (NADH) to NdmB for oxygen activation ((B)).
Therefore, in this study, we tried to clone the ndmB and ndmD genes in a pET32a vector and co-expressed NdmB and NdmD in recombinant BL21/pET32a. Then we purified the enzymes by Ni affinity chromatography. In addition, we demonstrated the catalytic activity of N-demethylases converting TB to 7-MX in the recombinant E. coli with ndmB and ndmD genes.
Materials and methods
Chemicals and reagents
Theobromine and agarose gel were purchased from Sigma–Aldrich (St. Louis, MO, USA). Tryptone, yeast extract, agar and Luria–Bertani broth (LB) were obtained from Becton Dickinson and Company (Sparks, MD, USA). NADH, isopropyl-D-thiogalactopyranoside (IPTG), 5-bromo-4-chloro-3-indolyl-D-galactopyranoside (X-Gal), and Tris base were obtained from RPI Corp. (Mt. Prospect, IL, USA). Restriction enzymes and ligation enzymes were purchased from New England BioLabs (Ipswich, MA, USA). Pfu Ultra DNA polymerase (Stratagene, Santa Clara, CA, USA), Taq DNA polymerase and Phusion HF polymerase (both from New England BioLabs) were used in various polymerase chain reactions (PCRs) as indicated. PCR primers were purchased from Integrated DNA Technologies (Coralville, IA, USA). PCR templates of pET32a/NdmBa3b2c1 and pET28/NdmD#S containing ndmB and ndmD genes from P. putida CBB5, respectively, were donated by the Center for Biocatalysis and Bioprocessing, University of Iowa, Coralville, IA. High-pressure liquid chromatography (HPLC)-grade methanol (J.T. Baker, Phillipsburg, NJ, USA) was used in all chromatographic studies.
Cloning of the ndmB, ndmD and ndmBHHD genes
To construct the NdmB-His and His-NdmD co-expression plasmid, the ndmB and ndmD genes were amplified by PCR. The designed PCR primers are shown in . The forward primer B-F-NdeI as well as the reverse primer BHHD-NR was used for PCR amplification of the ndmB genes with pET32a/NdmBa3b2c1 as the template. The forward primer BHHD-CF as well as the reverse primer D-R-BamHI was used for PCR amplification of the ndmD genes with pET28/NdmD#S as the template. The PCR reaction mixture included 0.5 μL of DNA template (containing about 50 ng), 5 nmol/L deoxyribonucleoside triphosphates (dNTPs) and 1× Phusion buffer (containing of MgCl2), 0.5 units of Phusion HF DNA polymerase and 12.5 pmol/L of each of the forward and reverse primers in a 25-μL final volume. The PCR amplification program (Thermo Electron PCR SPRT001 Issue3, Thermo Electron Corporation, Wilmington, NC, USA) included 35 cycles as follows: 98 °C for 10 s, 58 °C for 30 s and 72 °C for 60 s. The PCR amplification products of the ndmB and ndmD genes were mixed as templates for the overlap extension PCR according to Bryksin and Matsumura's procedure [Citation22]. To amplify the BHHD gene, PCR was conducted for 35 cycles with chimeric primers (B-F-NdeI, D-R-BamHI) and Phusion HF DNA polymerase as follows: 98 °C for 10 s, 58 °C for 30 s and 72 °C for 90 s. After purification, the PCR product was used as the insert gene fragment. The insert and the pET32a plasmid were separately incised by NdeI and BamHI to generate cohesive ends. The mixtures were incubated at 37 °C for 14 h to fully react. The incised inserts and plasmid were purified by the PCR clean-up kit, respectively. After that, the inserts and the plasmid containing the same cohesive ends were ligated at 16 °C for 16 h with T4 DNA ligase.
Table 1. PCR primers used in this study.
Then the purified product was transferred into electrocompetent E. coli DH5α cells (Invitrogen, Carlsbad, CA, USA) by electroporation. The pBHHD plasmid was designed to produce fusions of His-tag sequences to detect and purify the target protein. DNA sequencing analysis (Integrated DNA Technologies, Coralville, IA) confirmed the accuracy of the insertion fragment and BHHD.
Transformation of E. coli strain BL21 (DE3) and co-expression of BHHD
Expression studies were performed in E. coli BL21 (DE3). Transformation was carried out in the light of the pET System Manual (10th ed., TB055, Novagen, 2002). The pBHHD vectors were transformed. A colony of cells was transferred in 2 mL LB medium (ampicillin concentration: 100 μg/mL) and incubated at 37 °C for 8–10 h. After that, the cell suspension was transferred into 50 mL LB medium containing 100 μg/mL ampicillin and was cultured in a shaker/incubator at 250 r/min at 37 °C until an optical density at 600 nm (OD600) of 0.5 was reached. Then the culture was shifted to 18 °C in the presence of aseptic FeCl3 (work concentration of 10 μmol/L). Gene expression was induced with the help of IPTG (work concentration of 0.1 mmol/L) as the OD600 increased to 0.8–1.0. Sonication and cell disintegration was performed in an ice bath. The expression of the target genes was reflected by the total cell protein (TCP) stained by Coomassie blue. Therefore, sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) was employed to quantify the TCP.
Purification of NdmB and NdmD
Five grams of cryopreserved cells were unfrozen and suspended in potassium phosphate (KPi) buffer (pH 7.5, 25 mmol/L) which contained 10 mmol/L imidazole as well as 300 mmol/L NaCl. The cell suspension was lysed in a French press in freezing conditions at 138 MPa and the procedure was repeated once more. The lysed product was centrifuged at 30,000 ×g for 20 min. The supernatant containing the cell extracts was kept for the pBHHD purification procedure. The enzyme was purified at 4 °C using an automated fast protein liquid chromatography system (AKTA FPLC system; Amersham Pharmacia Biotech, Piscataway, NJ, USA). The soluble enzyme in the supernatant was passed through a Ni-NTA column (GE Healthcare, Pittsburgh, PA, USA) and purified at a speed of 3 mL/min. The column had a 17 mL-bed-volume and was pre-equilibrated by binding buffer (25 mmol/L KPi buffer (pH 7.0) containing 300 mmol/L NaCl and 10 mmol/L imidazole). Twenty milligrams of cell extract were passed through the Ni-NTA column and NdmB and NdmD were bound on the column for its His-tag. Binding buffer (100 mL) was used to wash the unbound protein off. The bound protein was eluted from the column by 60 mL elution buffer (KPi buffer (25 mmol/L, pH 7.5) containing 300 mmol/L NaCl and 250 mmol/L imidazole). The targeted protein was then concentrated by Amicon ultrafiltration units which were able to cut off the proteins of less than 10 kDa. The solution of concentrated enzyme was washed three times using 50 mmol/L KPi buffer (pH 7.5) and was centrifuged at 14,000 ×g and 4 °C for 30 min to remove imidazole with Amicon ultrafiltration (MWCO, 10 kDa). The purified enzymes were kept on ice for short-time storage or at −80 °C for long-term storage.
Enzyme activity and resting cell assays
The enzyme activity assay system included TB, NADH, Fe(NH4)2(SO4)2, as well as a proper amount of crude extract of pBHHD or purified concentrated enzyme solution in KPi buffer (50 mmol/L, pH 7.5), with a total volume of 1 mL. The concentrations of TB, NADH and Fe(NH4)2(SO4)2 were 0.5 mmol/L, 0.5 mmol/L and 50 μmol/L, respectively. The reaction mixture was incubated at 30 °C and was shaken on a microplate shaker/incubator (VWR, Radnor, PA) at 300 r/min. Then, 100 μL of sample was removed from the system at a fixed time and was mixed with equal-volume acetonitrile to block the reaction. The TB concentrations were quantified by HPLC. One unit of Ndms activity was equal to 1 μmol/min TB consumption.
To perform resting cell assays, the tested cells were centrifuged at 10,000 ×g for 20 min at 4 °C, washed twice with 50 mmol/L cold KPi buffer (pH 7.5) and then suspended in 50 mmol/L KPi buffer (pH 7.5). The reactions were carried out in 2 mL microcentrifuge tubes with a 1-mL total reaction volume containing an initial TB concentration of 1000 μmol/L. The suspensions were shaken at 30 °C and 300 r/min. Samples (100 μL) were taken periodically for HPLC analysis, and the concentration of TB in the reaction mixtures was calculated using a standard TB solution. Each measurement was carried out in three replicates.
Analytical procedures
Methylxanthines and their metabolites were identified and quantified with an HPLC system (Shimadzu LC-20AT, Tokyo, Japan) with an SPD-M20A photodiode array detector as well as a Hypersil BDS C18 column (4.6 × 125 mm) as described previously [Citation9]. The mobile phase was prepared as follows: MeOH:H2O:acetic acid at a ratio of 7.5:92.5:0.5. The flow rate was 0.5 mL/min. The protein concentration was quantified by the Bradford method and bovine serum albumin was employed as a protein level standard reacting with the dye reagent (Bio-Rad, Hercules, CA, USA) [Citation23].
Results and discussion
Cloning of ndmBHHD genes and analysis of recombinant pET32a-BHHD plasmid
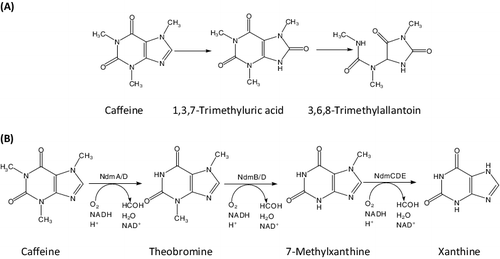
The ndmB and ndmD genes were amplified from pET32a/NdmBa3b2c1 and pET28/NdmD#S separately. The ndmBHHD gene was amplified by overlap extension PCR from the mixture of the PCR products. The PCR products were analyzed in an agarose gel (0.7% w/v) which displayed bands with the correct sizes. The bands corresponding to ndmB, ndmD and ndmBHHD were 1.1, 1.8 and 2.9 kb, respectively ((A)).
Figure 2. Electrophoretic separation (A) of the amplified NdmB, NdmD and NdmBHHD genes and analysis by restriction enzyme digestion (B) of recombinant pBHHD plasmid in 0.7% (w/v) agarose gel. (A) Lane 1, DNA ladder; Lane 2, NdmB gene (∼1.1 kb); Lane 3, NdmD gene (∼1.8 kb); Lanes 4–6, NdmBHHD genes (∼2.9 kb). (B) Lane 1, DNA ladder (Invitrogen, Carlsbad, CA, USA); Lane 2, recombinant plasmid incised by BamHI (8.3 kb); Lane 3, recombinant plasmid incised by BamHI and NdeI (1.8 and 6.5 kb); Lane 4, recombinant plasmid incised by BamHI and XbaI (2.9 and 5.4 kb); Lane 5, recombinant plasmid incised by NdeI and XbaI (1.1 and 7.2 kb).
The PCR product was cloned in the pET32a plasmid. Colonies containing plasmid with the recombinant BHHD gene were confirmed by PCR. The recombinant pBHHD plasmid was successfully incised by BamHI, BamHI and NdeI, BamHI and XbaI or NdeI and XbaI. The digested bands were extracted and transferred into a pET32a expression vector. The presence of the inserted gene was confirmed by PCR. The PCR primers were designed according to the sequence of pET32a and the BHHD gene and the resulting pBHHD plasmid was digested by NdeI and BamHI, BamHI and XbaI or NdeI and XbaI ((B)). Finally, the integrity and orientation of BHHD in the recombinant plasmid were confirmed by DNA sequencing.
Co-expression and purification of NdmB and NdmD
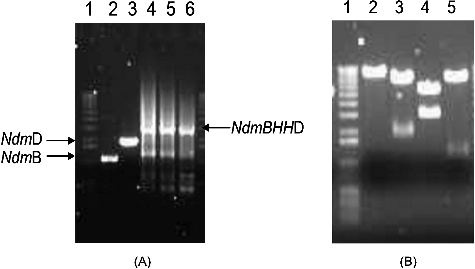
A plasmid containing the ndmB and ndmD genes was placed under the control of the T7 promoter in vector pET32a. E. coli BL21(DE3) harbouring BHHD expressed NdmB and NdmD at the same time when the cells were induced by IPTG. As shown in , the molecular mass of the two major protein bands corresponding to NdmB and NdmD was approximately 35 and 60 kDa, respectively. The results are consistent with previous studies reporting molecular mass of 35 and 65 kDa for NdmB and NdmD, respectively [Citation12,Citation13]. The purification of NdmB and NdmD from pBHHD was conducted by immobilized metal ion affinity chromatography according to a one-step purification method. The obtained results showed that the Ndms yield was 33.6% with a 10.3-fold purification (). Co-expression of ndmB and ndmD led to the expression of functional NdmB activity.
Figure 3. SDS-PAGE analysis of the expression of recombinant proteins. Lane 1, protein molecular weight marker (14–191 kDa; Invitrogen, Carlsbad, CA, USA); Lane 2, Ni-column load; Lane 3, flow-through; Lane 4, wash; Lane 5, elution of NdmB and NdmD. Coomassie brilliant blue R-250 staining.
Table 2. Purification of Ndms from recombinant BL21/pET32a BHHD.
Resting cell assay
Summers et al. [Citation24] constructed a recombinant E. coli with ndmA and ndmD that was able to effectively convert caffeine to TB. Algharrawi et al. [Citation21] reported the direct conversion of TP to 3-MX by metabolically engineered E. coli with ndmA and ndmD (pDdA), yielding 100 mg of highly pure 3-MX from TP. This microbial conversion provided an economical, environmentally friendly approach to produce these chemicals in large quantities. In order to demonstrate whether our engineered cells have N3-demethylation catalytic activity, we further performed resting cell assay using TB as a reaction substrate. As shown in , 1000 μmol/L TB was completely transformed into 7-MX within 90 min under the reaction conditions of 30 °C, pH 7.5, 300 r/min at a final cell density of OD600 = 5. This demonstrated that the recombinant pBHHD cells had a high biocatalytic capacity for N-demethylation, which may provide a promising producer of 7-MX from TB after optimization of the process in subsequent experiments.
Figure 4. Stoichiometric N-demethylation of TB catalyzed by recombinant pBHHD cell. Reaction conditions: 50 mmol/L KPi (pH 7.5), T = 30 °C, 300 r/min. Total volume: 1 mL, [TB] = 1000 µmol/L; final OD600 = 5.
![Figure 4. Stoichiometric N-demethylation of TB catalyzed by recombinant pBHHD cell. Reaction conditions: 50 mmol/L KPi (pH 7.5), T = 30 °C, 300 r/min. Total volume: 1 mL, [TB] = 1000 µmol/L; final OD600 = 5.](/cms/asset/f61fe8a8-31d9-4fac-9979-2c41d6a4c81e/tbeq_a_1295819_f0004_b.gif)
Conclusions
In this study, ndmB and ndmD were cloned together in a recombinant pET32a-BHHD plasmid, electroporated into E. coli strain BL21 (DE3) and co-expressed successfully. Co-expressed NdmB and NdmD were purified using Ni-affinity chromatography with 10.3-fold purification and 33.6% yield. The purification step was efficient and simple. The enzyme activity of NdmB and NdmD showed that 1000 μmol/L TB was converted completely into 7-MX in 90 min by resting cells harbouring pBHHD. These results indicate that this may be a promising approach to produce 7-MX from TB, which could be explored in further studies.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Gummadi SN, Bhavya B. Enhanced degradation of caffeine and caffeine demethylase production by Pseudomonas sp. in bioreactors under fed-batch mode. Appl Microbiol Biotechnol. 2011;91:1007–1017.
- Sara ML, Beatriz S, Miren GJ , et al. Theobromine, caffeine, and theophylline metabolites in human plasma and urine after consumption of soluble cocoa products with different methylxanthine contents. Food Res Int. 2014;63:446–455.
- Beltran JG, Leask RL, Brown WA. Activity and stability of caffeine demethylases found in Pseudomonas putida IF-3. Biochem Eng J. 2006;31:8–13.
- Gopishetty SR, Louie MT, Yu CL , et al. Microbial degradation of caffeine, methylxanthines and its biotechnological applications. In: Thatoi HN Mishra BB, editors. Microbial biotechnology: method and applications. Oxford (UK): Alpha Sci Int Ltd. Press; 2011. p. 44–67.
- Arnaud MJ. Pharmacokinetics and metabolism of natural methylxanthines in animal and man. In: Fredholm BB, editor. Methylxanthines. Berlin: Springer-Verlag; 2011. p. 33–91. ( Handbook of experimental pharmacology; vol. 200).
- Brand D, Pandey A, Leon JR , et al. Relation between coffee husk caffeine degradation and respiration of Aspergillus niger in solid state fermentation. Appl Biochem Biotechnol. 2002;102:168–178.
- Mohapatra BR, Harris N, Nordin R , et al. Purification and characterization of a novel caffeine oxidase from Alcaligenes species. J Biotechnol. 2006;125:319–327.
- Yu CL, Kale Y, Gopishetty S , et al. A novel caffeine dehydrogenase in Pseudomonas sp. strain CBB1 oxidizes caffeine to trimethyluric acid. J Bacteriol. 2008;190:772–776.
- Yu CL, Louie TM, Summers RM , et al. Two distinct pathways for metabolism of theophylline and caffeine are coexpressed in Pseudomonas putida CBB5. J Bacteriol. 2009;191:4624–4632.
- Madyastha KM, Sridhar GR. A novel pathway for the metabolism of caffeine by a mixed culture consortium. Biochem Biophys Res Commun. 1998;249:178–181.
- Summers RM, Louie TM, Yu CL , et al. Characterization of a broad-specificity non-haem iron N-demethylase from Pseudomonas putida CBB5 capable of utilizing several purine alkaloids as sole carbon and nitrogen source. Microbiology. 2011;157:583–592.
- Summers RM, Louie TM, Yu CL , et al. Novel, highly specific N-demethylases enable bacteria to live on caffeine and related purine alkaloids. J Bacteriol. 2012;194:2041–2049.
- Summers RM, Seffernick JL, Quandt EM, et al. Caffeine Junkie: an unprecedented glutathione S-transferase-dependent oxygenase required for caffeine degradation by Pseudomonas putida CBB5. J Bacteriol. 2013;195:3933–3939.
- Yu CL, Summers RM, Li YL , et al. Rapid identification and quantitative validation of a caffeine degrading pathway in Pseudomonas sp. CES. J Proteome Res. 2015;14:95–106.
- De Sena AR, de Assis SA, Branco A. Analysis of theobromine and related compounds by reversed phase high-performance liquid chromatography with ultraviolet detection: an update (1992–2011). Food Technol Biotechol. 2011;49:413–423.
- Trier K, Madsen SMR, Cui DM , et al. Systemic 7-methylxanthine in retarding axial eye growth and myopia progression: a 36-month pilot study. J Ocul Biol Dis Infor. 2008;1:85–93.
- Dash SS, Gummadi SN. Catabolic pathways and biotechnological applications of microbial caffeine degradation. Biotechnol Lett. 2006;28:1993–2002.
- Gummadi SN, Bhavya B, Ashok N. Physiology, biochemistry and possible applications of microbial caffeine degradation. Appl Microbiol Biotechnol. 2012;93:545–554.
- Jin L, Bhuiya MW, Li MM , et al. Metabolic engineering of Saccharomyces cerevisiae for caffeine and theobromine production. PLoS ONE. 2014;9(8):e105368.
- Summers RM, Mohanty SK, Sridhar GR , et al. Genetic characterization of caffeine degradation by bacteria and its potential applications. Microb Biotechnol. 2015;3:369–378.
- Algharrawi KHR, Summers RM, Sridhar GR , et al. Direct conversion of theophylline to 3-methylxanthine by metabolically engineered E. coli. Microb Cell Fact. 2015;14:203–214.
- Bryksin AV, Matsumura I. Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. Biotechniques. 2010;48:463–465.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem. 1976;72:248–254.
- Summers RM, Sridhar GR, Mohanty SK , et al. New genetic insights to consider coffee waste as feedstock for fuel, feed, and chemicals. Cent Eur J Chem. 2014,1:1271–1279.