
ABSTRACT
MIP is a candidate tumour suppressor protein found in hepatocellular carcinoma. It can interact with different proteins in different subcellular locations. However, antibodies against MIP for testing purposes are still not available and this has hindered the research on MIP protein. In this study, the full coding sequence of MIP protein was cloned into the prokaryotic expression vector pET-28a. The recombinant protein his-MIP was expressed by IPTG (isopropyl β-D-1-thiogalactopyranoside) induction and purified by nickel-affinity chromatography, then used to immunize New Zealand rabbits to obtain a polyclonal antibody against MIP. The sensitivity and specificity of the antibody were evaluated by enzyme-linked immunosorbent assay and western blot. We used the obtained antibody to detect endogenous MIP protein in hepatocellular carcinoma (HCC) cells and normal human hepatic cells L-O2 and found decreased MIP expression in HCC cells. The immunohistochemistry results further showed that the expression level of MIP protein in HCC tissues was lower than that in the corresponding adjacent non-cancerous tissues. The MIP protein was distributed mainly in the cytoplasm in HCC. In adjacent tissues, however, high expression of MIP was detected in the nucleus in addition to the cytoplasm. The prepared polyclonal antibody against MIP provides a useful tool for further research on the functions of MIP.
Introduction
MIP (MafF interacting protein, also known as MAFIP, pp5644) is a tumour-associated protein first discovered in human hepatocellular carcinoma (HCC) by subtractive hybridization in 2004 [Citation1]. The mRNA expression profile analysis has shown that MIP is expressed in human tissues such as liver, brain, kidney, heart and lung; but it is absent in skeletal muscle and pancreas tissues [Citation2]. Compared with the corresponding normal tissues, the mRNA level of MIP is decreased significantly in some tumours [Citation3]. MIP overexpression inhibits the growth and colony formation of human HCC cell line SMMC7721 [Citation4]. Moreover, exogenous overexpression of MIP in Hela cells can inhibit tumour formation in nude mice [Citation3]. Therefore, MIP is regarded as a candidate tumour suppressor protein.
To date, three MIP-interacting proteins have been identified: hMis18α (MIS18 kinetochore protein A), NEMO (NFκB essential modulator) and MafF (MAF bZIP transcription factor F). Cellular immunofluorescence assay in Hela cells has shown that MIP is located in the cytoplasm with punctate distribution [Citation2]. MIP can interact with hMis18α and NEMO in the cytoplasm. However, when co-transfected with MafF, which is localized in the nucleus [Citation5], MIP can shuttle from the cytoplasm into the nucleus and interact with MafF, acting as a co-activator of MafF to activate transcription of specific DNA cis-elements [Citation6]. However, research on MIP confronts many restrictions due to the lack of a commercial antibody against MIP. Therefore, the preparation of MIP antibody is critical for further research on the expression, localization and function of the MIP protein.
The aim of this study was to prepare a specific antibody against MIP for evaluating the expression and subcellular localization of MIP protein in HCC.
Materials and methods
Strains, cells, vector and reagents
Escherichia coli Top10 and BL21 (DE3) strains, pCMV-HA2-MIP plasmid and prokaryotic expression vector pET-28a were stored at the Department of Histology and Embryology, Guangdong Medical University. Human HCC cell lines HepG2, SMMC7721, SK-Hep1 and normal hepatic cells L-O2 were purchased from the Cell Bank of the Chinese Academy of Sciences. Restriction endonucleases, DNA polymerase and T4 DNA ligase were purchased from Takara (Dalian, China). Isopropy-β-D-thiogalactoside (IPTG), complete and incomplete Freund's adjuvants and monoclonal antibody against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were purchased from Sigma–Aldrich (Munich, Germany). The HisPur Ni-NTA Spin Purification Kit, M-PER Mammalian Protein Extraction Reagent, horseradish peroxidase (HRP)-conjugate goat anti-rabbit IgG and Fast ECL Western blotting kit were purchased from Thermo Fisher (Waltham, USA). The Streptavidin-Peroxidase Detection Kit for immunohistochemistry was purchased from ZSGB Bio (Beijing, China).
Animals and clinical tissue samples
Female adult New Zealand rabbits were purchased from the Guangdong Medical Laboratory Animal Center. Twenty paired samples of HCC and the corresponding adjacent non-cancerous tissues were obtained from the Affiliated Hospital of Guangdong Medical University. All of the patients gave their signed informed consent. The project was approved by the local institutional ethics council.
Plasmid construction
Based on the known MIP cDNA sequence and multiple cloning sites of pET-28a vector, primers for amplifying the MIP open reading frame (ORF) were designed with the Primer 5.0 software. The sense primer was 5’-CGCCATATGCTCTGCCCTAGAGCCG-3’, containing a NdeI site (underlined). The antisense primer was 5’-GACTCGAGCTGGTAGGCAGCCAGC-3’, containing a XhoI site (underlined). The DNA sequence encoding human MIP protein was amplified by polymerase chain reaction (PCR) from the pCMV-HA2-MIP plasmid constructed previously [Citation2]. The purified PCR product was digested with XhoI and NdeI, and then ligated to pretreated pET-28a vector digested with the same two enzymes. The recombinant plasmid pET-28a-MIP was confirmed by double restriction enzyme digestion and DNA sequencing.
Expression and purification of his-MIP recombinant protein
The recombinant plasmid pET-28a-MIP was transformed into E. coli BL21 (DE3). Single colonies from transformants were transferred to 5 mL of Luria–Bertani medium supplemented with 100 μg/mL ampicillin and incubated at 37 °C overnight. This culture was diluted 1:100 into 500 mL of fresh medium and the incubation continued. When the culture reached an optical density at 600 nm (OD600) of 0.5–0.6, the expression of the his-MIP recombinant protein was induced by 0.4 mmol/L IPTG with incubation for 6 h at 28 °C. The induced bacteria were harvested by centrifugation at 5000 × g for 10 min at 4 °C. The pellet was re-suspended in ice-cold phosphate buffered saline (PBS) at a ratio of 5 mL buffer per 1 g cell pellet. Following sonication, the suspension was centrifuged at 12,000 × g for 10 min and the supernatant was collected. A volume of 1 mL of induced or non-induced bacteria culture was centrifuged and the pellet was resuspended in 100 μL of PBS. An equal volume of each sample was mixed with 2 × sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer and subjected to 12% SDS-PAGE electrophoresis. The gels were stained with Commassie blue to detect the expression of the his-MIP recombinant protein. Then the supernatant was transferred into a tube containing a Ni-NTA Spin column. The column was washed three times with washing buffer (100 mmol/L NaH2PO4, 10 mmol/L Tris/HCl and 10 mmol/L imidazole, pH 8.0). The bound his-MIP proteins were eluted from Ni-NTA Spin with elution buffer (100 mmol/L NaH2PO4, 10 mmol/L Tris/HCl and 250 mmol/L imidazole, pH8.0). The flow-through and the eluent were analysed by SDS-PAGE.
Animal immunization
Routine immunization protocols were used to generate the polyclonal antibody against MIP [Citation7]. Briefly, the rabbits were injected subcutaneously with immunogen six times at two-week intervals. At the first injection, 500 μg of his-MIP fusion protein mixed with Freund's complete adjuvant at a ratio of 1:1 (v/v) was used to immunize the rabbits. In the subsequent five booster injections, 250 μg of his-MIP protein in Freund's incomplete adjuvant at 1:1 (v/v) was used. Two weeks after the last immunization, the antisera were harvested. Rabbit sera before the first immunization were collected and used as negative controls.
Enzyme-linked immunosorbent assay (ELISA) analysis of antisera
The his-MIP recombinant protein was diluted to 5 μg/mL in carbonate buffer and coated onto 96-well high binding ELISA plates with 100 μL per well. The plates were incubated overnight at 4 °C. After washing three times with PBS containing 0.1% Tween 20 (PBST), the plates were blocked with 200 μL blocking buffer (PBST and 1% bovine serum albumin) in each well for 2 h and were washed three times with PBST. The antisera were diluted in blocking buffer (1:3125, 1:6250, 1:12,500, 1:25,000, 1:50,000), and 100 μL of diluted antisera were transferred into the ELISA plate. An aliquot of 100 μL of preimmune sera (1:1000 dilution) was used as negative controls. The plates were incubated at room temperature for 1 h and then washed. The HRP-labelled goat anti-rabbit IgG (1:5000 dilution, 100 μL per well) was added, and the plate was incubated at 37 °C for 45 min. After washing four times, 100 μL 3,3,5,5-tetramethylbenzidine was added to each well, followed by incubated in a darkroom at 37 °C for 10 min. The absorbance at 450 nm was measured with a microplate reader (Multiskan MK3, Thermo, Waltham, USA).
Western blot assays of the recombinant and endogenous MIP protein
A volume of 1 mL of bacteria culture (non-transformed, non-induced or induced) was centrifuged at 5000 × g for 10 min. The bacteria pellet was resuspended in 100 μL of PBS and 100 μL of 2 × SDS-PAGE loading buffer. The total protein of cultured human hepatic cells was extracted using M-PER Mammalian Protein Extraction Reagent according to the manufacturer's instructions. The lysed bacteria or the total protein of human hepatic cells were subjected to SDS-PAGE. After electrophoresis, proteins were transferred to a polyvinylidene difluoride membrane at 140 mA for 90 min. After washing with PBST, the membrane was blocked with 5% non-fat milk in PBST buffer for 2 h and then incubated overnight at 4 °C with the anti-MIP antibody (1:2000 dilution). Anti-GAPDH antibody (1:20,000 dilution) was used as an internal control. The membrane was washed three times with PBST and incubated with HRP-conjugated goat anti-rabbit IgG (1:5000 dilution) for 2 h at room temperature. After washing with PBST, the blot was visualized with an ECL detection kit.
Immunohistochemistry assay
Immunohistochemistry was conducted by the streptavidin–peroxidase method. Sections of tissue samples from patients were deparaffinized with xylene and rehydrated. For antigen retrieval, sections were immersed in citrate buffer and microwaved for 5 min. To eliminate inherent peroxidase activity, sections were incubated with 3% H2O2 for 15 min. Sections were incubated with the anti-MIP polyclonal antibody (1:200 dilution) overnight at 4 °C. After washing with PBS, the sections were incubated with the biotin-labelled secondary antibody for 15 min and then incubated with HRP-labelled streptavidin for 15 min, respectively. Signals were visualized with 3,3'-diaminobenzidine for 1 min, with slight counterstaining with hematoxylin. For negative controls, the primary antibody was replaced with PBS in each experiment. The sections were evaluated independently by two investigators as previously described [Citation8]. Briefly, the degree of immunohistochemical staining was evaluated by multiplying the staining intensity score (0: no, 1: light yellow, 2: yellow, 3: brown yellow) and the staining proportion score (0: <25%, 1: 25%–50%, 2: 51%–75%, 3: >75%). Tissues with a final score of 4–9 were regarded as positive.
Statistical analysis
Data were analysed by SPSS 13.0 software. One-way analysis of variance (ANOVA) was used in western blot analysis and Fisher's exact test was used for analysing the results of immunohistochemistry assays. P < 0.05 was regarded as statistically significant.
Results and discussion
Construction and identification of pET-28a-MIP recombinant
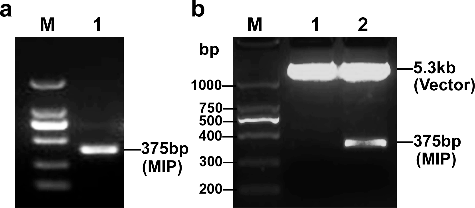
The full-length open reading frame of MIP is composed of 375 base pairs (bp), encoding a 124-amino acid (aa) protein. This coding sequence was obtained by PCR amplification. (a) shows that the size of the PCR product was approximately 375 bp. The double-enzyme digestion of the recombinant plasmid demonstrated that the gene fragment was successfully inserted into the pET-28a prokaryotic expression vector ((b)). DNA sequencing confirmed that the sequence and the reading frame of the inserted fragment were correct (data not shown).
Figure 1. Construction and identification of the pET-28a-MIP recombinant plasmid. (a) PCR amplification to obtain the full-length ORF of the MIP protein. Lane M, DNA ladder (Takara, Dalian, China); lane 1, PCR product. (b) Recombinant plasmids identified by double restriction enzyme digestion. Lane M, DNA ladder (Takara, Dalian, China); lane 1, pET-28a vector; lane 2, pET-28a-MIP recombinant plasmid.
Expression and purification of his-MIP protein
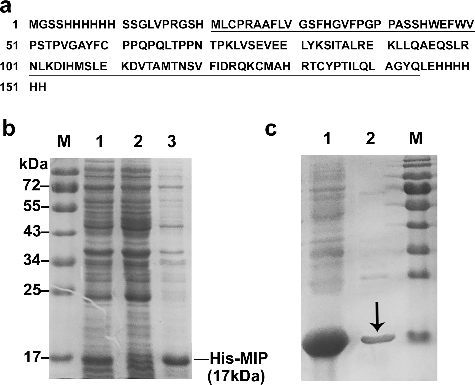
After transformation with pET-28a-MIP, E.coli BL21(DE3) cells were induced at 28 °C for 6 h with 0.4 mmol/L IPTG to express the recombinant protein his-MIP (152 aa, (a)), which contains the full MIP protein sequence (124 aa, underlined in (a)) and His-tags at both the N-terminus and C-terminus. The whole lysate of induced or non-induced bacteria, and the supernatant of induced bacteria after sonication were analysed by SDS-PAGE, respectively. The results showed that the expression efficiency was promoted after IPTG induction and the content of his-MIP protein, with the expected molecular weight of about 17 kDa, was increased significantly (lane 1 in (b)) compared with the non-induced sample (lane 2 in (b)). In the supernatant of induced bacteria lysate, there was a strong band at 17 kDa which indicated soluble his-MIP protein (lane 3 in (b)). Therefore, the supernatant was collected and the his-MIP recombinant proteins were purified using Ni-NTA Spin affinity column. After elution with imidazole, the electrophoresis result showed a clear 17 kDa band in the eluant ((c)).
Figure 2. Expression and purification of the his-MIP recombinant protein. (a) Amino-acid sequence of the his-MIP recombinant protein (152 aa), which contains the full amino acid sequence of the native MIP protein (124 aa, underlined). (b) Expression of his-MIP induced by IPTG in E.coli BL21 (DE3) analysed by Coommassie blue staining after 12% SDS-PAGE. Lane M, protein marker; lane 1, whole lysate of induced bacteria; lane 2, whole lysate of non-induced bacteria; lane 3, supernatant of induced bacteria after sonication. (c) Purification of his-MIP using Ni-NTA Spin column. Lane 1, supernatant before purification; lane 2, eluant after purification; lane M, protein size marker (Genview, El Monte, USA); black arrow, purified his-MIP protein.
Preparation and characterization of the anti-MIP polyclonal antibody
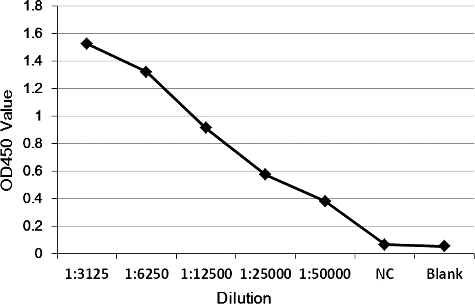
After six immunizations of rabbits using purified his-MIP recombinant protein, the antisera were harvested and analysed by ELISA. The results showed that the titre of the antisera was more than 1:50,000 ().
Figure 3. Titre determination of the antisera by ELISA.
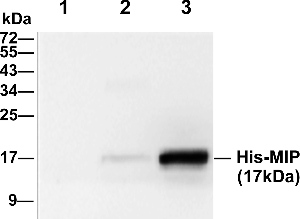
In order to evaluate the specificity of the anti-MIP polyclonal antibody, we used prepared rabbit antisera as the primary antibody to detect his-MIP protein in the whole lysate of non-transformed bacteria, non-induced and induced bacteria by western blot. shows that there was no signal in non-transformed bacteria, but there was a weak signal in lysed non-induced bacteria with the expected MW of 17 kDa. Moreover, a clear and strong band was detected at the same 17 kDa position in the lysed induced bacteria, which indicated the specific signal of his-MIP recombinant protein. The western blot results demonstrated that the prepared antibody has high specificity.
Figure 4. Evaluation of the specificity of the anti-MIP antibody by western blot. Lane 1, whole lysate of non-transformed bacteria; lane 2, whole lysate of non-induced bacteria; lane 3, whole lysate of induced bacteria.
Expression of endogenous MIP protein in normal hepatic cells and HCC cells
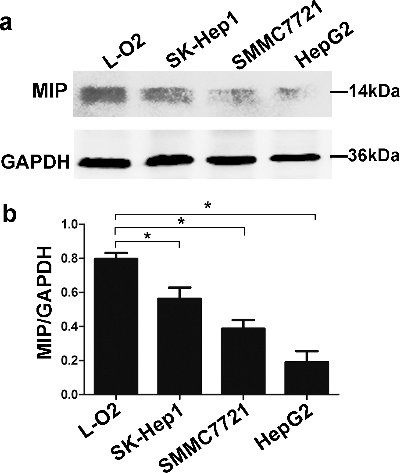
The expression of endogenous MIP protein in three HCC cell lines and normal hepatic cell L-O2 was analysed by western blot using the prepared antibody. In all of the above cells, there was a clear band about 14 kDa, which is consistent with the molecular size of native MIP protein (124 amino acids). The results also showed that the expression level of MIP protein was decreased in HCC cell lines compared to normal human hepatic cell L-O2 (). The experiments verified that the antibody could detect endogenous MIP protein in cells.
Figure 5. Expression of endogenous MIP protein in normal human hepatic cell line L-O2 and three HCC cell lines, detected by Western blot using prepared anti-MIP antibody. (a) Total proteins of cultured cells extracted using Mammalian Protein Extraction Reagent. (b) Comparison of the expression level of MIP protein in human normal hepatic cells and HCC cells based on MIP/GAPDH ratio.
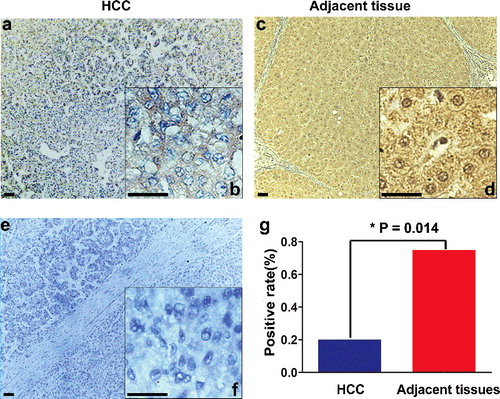
Expression of MIP protein in HCC tissues
The expression and distribution of endogenous MIP protein in HCC tissues were analysed immunohistochemically using the prepared antibody. There was a significant difference in the expression pattern of MIP protein between the HCC tissues ((a,b)) and the corresponding adjacent tissues ((c,d)). (g) shows that the positive MIP expression rate in HCC tissues (4/20, 20%) was lower than that in the adjacent tissues (15/20, 75%), P < 0.05. Moreover, the subcellular distribution of MIP protein also differed: it was mainly distributed unevenly in the cytoplasm in HCC tissues ((b)), but there was significant MIP expression in the nucleus in addition to the cytoplasm in adjacent tissues ((d)).
Figure 6. Immunohistochemical staining of MIP protein in HCC and corresponding adjacent tissues. (a,b) Weak cytoplasmic expression of MIP in HCC tissue; (c,d) strong nuclear and cytoplasmic expression of MIP in the corresponding adjacent tissue; (e,f) negative control by replacing the anti-MIP primary antibody with PBS in the HCC tissue; (g) comparison of the positive MIP immunohistochemical staining rate in 20 paired samples of HCC and adjacent tissues.
As a candidate tumour suppressor protein, the distribution and expression level of MIP are worth studying. In western blot assays, the prepared antibody showed a high specificity to MIP protein. Using this antibody, we confirmed the expression of endogenous MIP protein in human hepatic cells and found decreased expression in HCC cells and tissues compared to normal hepatic cells and tissues. What is more, there was an obvious reduction in the nuclear distribution of MIP protein in HCC tissues compared with adjacent tissues. The tissue distribution and subcellular localization of proteins are critical to their functions [Citation9], such as NFκB [Citation10] and ERK [Citation11]. Combined with previous reports that MIP can shuttle between the cytoplasm and the nucleus and can interact with MafF in the nucleus to activate transcription [Citation6,Citation12], our findings suggested that MIP may participate in the transcriptional regulation of some HCC-related genes. In could be speculated that low expression level and lack of nuclear distribution of MIP protein may be related to the tumorigenesis of HCC.
Conclusions
MIP is a candidate tumour suppressor protein and can interact with multiple proteins. However, the lack of a specific antibody has limited the research on its function. In this study, a polyclonal antibody against MIP was prepared by immunizing rabbits with purified his-MIP fusion protein. The antibody could specifically recognize MIP protein and was successfully applied in western blot and immunohistochemical analysis. The novel antibody provides a useful tool for further research on the functions of MIP.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Wan D, Gong Y, Qin W, et al. Large-scale cDNA transfection screening for genes related to cancer development and progression. Proc Natl Acad Sci USA. 2004;101:15724–15729.
- Ye XX, Lu H, Yu Y, et al. P5644 interacts with phosphatidylinositol-4-phosphate adaptor protein-1 associated protein-1. Mol Cell Biochem. 2005;271:151–158.
- Li Y, Yu Y, Zhang Y, et al. MAFIP is a tumor suppressor in cervical cancer that inhibits activation of the nuclear factor-kappa B pathway. Cancer Sci. 2011;102:2043–2050.
- Ye X, Lu H, Huo K, et al. Finding a novel interacting protein of the hepatic carcinoma related gene MIP: NF-kappaB essential modulator (NEMO). Oncol Rep. 2011;25:231–235.
- Kannan MB, Solovieva V, Blank V. The small MAF transcription factors MAFF, MAFG and MAFK: current knowledge and perspectives. Biochim Biophys Acta. 2012;1823:1841–1846.
- Ye X, Li Y, Huang Q, et al. The novel human gene MIP functions as a co-activator of hMafF. Arch Biochem Biophys. 2006;449:87–93.
- Cooper HM, Paterson Y. Production of polyclonal antisera. Curr Protoc Neurosci. 2009;48: 5.5.1–5.5.10.
- Wu M, Ye X, Deng X, et al. Upregulation of metastasis-associated gene 2 promotes cell proliferation and invasion in nasopharyngeal carcinoma. Onco Targets Ther. 2016;9:1647–1656.
- Tiligada E. Nuclear translocation during the cross-talk between cellular stress, cell cycle and anticancer agents. Curr Med Chem. 2006;13:1317–1320.
- Kim SJ, Yoon S. Activated Rac1 regulates the degradation of IkappaBalpha and the nuclear translocation of STAT3-NFkappaB complexes in starved cancer cells. Exp Mol Med. 2016 [cited 2017 Mar 11];48:e231. DOI:10.1038/emm.2016.17
- Wainstein E, Seger R. The dynamic subcellular localization of ERK: mechanisms of translocation and role in various organelles. Curr Opin Cell Biol. 2016;39:15–20.
- Ye X, Huo K, Chen D. [Subcellular localization and changes of tumor-related gene MIP in Hela cells]. Acta Anat Sin. 2011;42:175–179. Chinese.