
ABSTRACT
Cardiovascular diseases (CVD) comprise a broad range of disorders of the heart and blood vessels. In this study, we used next-generation sequencing with a panel that includes 174 genes connected to CVD in order to investigate the possible genetic causes that underline some clinical phenotypes and their severity. Two patients were found with double heterozygosity, each carrying one new variant. One patient with supravalvular aortic stenosis has novel ELN: c.890-1G>A and a known variant SCN5A: p.Gly9Val in a heterozygous state, whereas another patient with hypertrophic cardiomyopathy has a heterozygous novel CACNA1C: p.Arg514Gly and a known SCN5A: p.Arg800His variant. This method proved to be useful in determining the mutation status in correlation with the severity of the clinical phenotype and can further clarify cases where the clinical status could not be explained only by single gene mutation detected by standard methods.
Introduction
Cardiovascular diseases (CVD) include disorders of the vasculature, the myocardium, the electrical circuit of the heart and congenital heart disease [Citation1]. They encompass a wide range of different clinical entities, including ischemic heart disease or coronary artery disease, cerebrovascular disease, diseases of the aorta and arteries including hypertension and peripheral vascular diseases, congenital heart disease, rheumatic heart disease, cardiomyopathies and cardiac arrhythmias [Citation2]. Furthermore, cardiomyopathies – as diseases of the myocardium associated with cardiac dysfunction – are classified into primary cardiomyopathies, i.e. disorders of the heart muscle due to genetic, non-genetic or acquired causes; and secondary cardiomyopathies, i.e. myocardial damage due to multi-organ or systemic disease [Citation3]. According to the World Health Organization's fact sheet reviewed in September 2016, CVD are the first cause of death worldwide with more than 17 million deaths annually. Over three quarters of this number are in low- and middle-income countries.
There is a genetic contribution to most cardiovascular diseases. Localization and detection of genes of monogenic forms of CVD connected to a single gene that leads to certain clinical phenotype has been done for decades by linkage analysis and direct DNA sequencing [Citation4]. The genetics of the human cardiovascular diseases from determined Mendelian forms of CVD and common, complex forms of heart disease to the novel techniques, such as next-generation approach and modeling human genetic diseases in reprogrammed cells, is reviewed by Kathiresan and Srivastava [Citation5].
With the advance of high-throughput technologies such as next-generation sequencing, cardiovascular genetic disorders have been associated with thousands of new variants in different genes introducing high prevalence of novel variants predominantly connected with mutations in single genes. Interestingly, the rate of double or compound heterozygosity is 3%–5% [Citation6]. Double heterozygosity as digenic inheritance (DI) is the simplest form of inheritance for genetically complex diseases; examples of human DI are reviewed in [Citation7]. Next-generation sequencing has simplified the detection of two genes connected to a phenotype of interest, continuing where Sanger sequencing left off in the past. By sequencing one gene at a time, scientists mostly stopped looking for mutations once one mutant gene was detected, although very often the correlation between the genotype and phenotype was inconclusive and possibly could have been explained clearly by a second gene involved.
We report two cases with double heterozygosity of newly detected variants in two distinctive genes connected to severe clinical phenotype randomly found by means of next-generation sequencing using a gene panel of 174 genes connected to cardiovascular diseases. Our aim was to correlate the different phenotypes in two patients carrying pathological variants in SCN5A with the involvement of a second pathological mutation in a different gene. One of the probands is diagnosed with supravalvular aortic stenosis (SVAS) with severe hypoplasia of both branches of the pulmonary artery and the other one is with sporadic form of asymmetric obstructive hypertrophic cardiomyopathy (HCM). Both were found to be double heterozygotes, namely the first patient has a novel ELN: c.890-1G>A and a known variant SCN5A: p.Gly9Val, while the second one has a novel CACNA1C: p.Arg514Gly and known SCN5A: p.Arg800His variant.
ELN (https://www.ncbi.nlm.nih.gov/gene/2006) is a gene of 34 exons that encodes a protein involved in the elasticity of various tissues and organs. The soluble monomer product of the gene is called tropoelastin and consists of a hydrophobic domain responsible for the elastic properties and lysine-rich sequences needed for lysyl oxidase-mediated covalent cross-linking between monomers making an insoluble network of elastic fibres in the extracellular space [Citation8] For review, see [Citation9].
Gene SCN5A (https://www.ncbi.nlm.nih.gov/gene/6331) encodes a member of the voltage-gated sodium channel family (Sodium Channel, voltage-gated, Type V, Alpha Subunit) responsible for initiating the cardiac action potential [Citation10]. This protein mediates the voltage-dependent permeability of the cell membranes for sodium ions. The inactivation of the channel is regulated by the intracellular calcium levels.
Gene CACNA1C (https://www.ncbi.nlm.nih.gov/gene/775) encodes the alpha1C subunit of the voltage-dependent calcium channels (Voltage-dependent L-type calcium channel subunit alpha-1C). Voltage-dependent calcium channels (VSCC) consist of a complex of alpha1, alpha2/delta, beta and gamma subunits in a 1:1:1:1 ratio and mediate the entrance of the calcium ions into the cells. These channels are involved in series of calcium-dependent processes including muscular contraction, secretion of hormones and neurotransmitters, gene expression, cell movement, cell division and cell death. Calcium channels that contain the alpha1C subunit play an important role in coupling the processes of excitation and reduction in the heart. Calcium ion entry into the cardiomyocyte via L-type calcium channel provides the initiating event in cardiac excitation-contraction (EC); therefore, even small perturbations of its function lead to profound cardiac diseases [Citation11].
Materials and methods
Clinical evaluation
The patients attended the National Heart Hospital – Sofia. Cardiac evaluation and diagnosis was performed according to the standard procedures for clinical examination in the hospital. After institutional approval, blood samples were obtained for DNA extraction from the patients and their parents. Informed consent was obtained from all individual participants included in the study.
Gene analysis
Genomic DNA was extracted from blood using a commercially available QIAamp DNA Blood Mini Kit (Qiagen). The genomic material was quantitatively and qualitatively assessed by Qubit dsDNA BR Assay Kit, Life Technologies and NanoDrop 2.0. Library preparation and enrichment was performed using the TruSight Cardio panel by Illumina, which includes 174 genes associated to cardio-vascular diseases. Quantification of the enriched DNA libraries was done by quantitative polymerase chain reaction (qPCR) on an Illumina qPCR Eco system using KAPA Library Quantification Kit. The reactions were run in triplicate so that accurate concentrations of the dilutions could be calculated. Denaturation and dilution of libraries was done according to the MiSeq System Denature and Dilute Libraries Guide. They were loaded onto a cartridge for sequencing as described in the MiSeq System User Guide (part # 150276). Sequencing was performed on a MiSeq System using the MiSeq Reagent Kit v2.
Sequencing data were analysed by Softgenetics NextGene Software (version 2.3.3). After alignment to the Human reference sequence – Genome Reference Consortium Human Build 37 (GRCh37/hg19), a list of variants was formed. Variant annotation and filtering was done using the VariantStudio Software. Variants in candidate genes were confirmed by Sanger sequencing.
Results and discussion
The first patient, a second child in the family, was diagnosed with peripheral pulmonary stenosis common for patients with Williams–Beuren syndrome but without other phenotypical traits characteristic of the syndrome and no familial history of cardiovascular diseases nor diseases of the connective tissue. Pulmonary artery stenosis is a narrowing that occurs either in the main pulmonary artery and/or in the left or right pulmonary artery branches. Since the pulmonary artery is a large vessel that delivers oxygen-poor blood from the right ventricle into the lungs to be enriched with oxygen, the narrowing causes a reduction in the volume of blood that reaches the lungs. The heart pumps harder in order to overcome the narrowing, the pressure in the right ventricle is increased, which may damage the heart muscle in this area.
At four months of age, heart murmur was detected and pulmonary stenosis with gradient 60 mmHg was diagnosed, without cyanosis and failure. Catheterization under general anaesthesia was performed. Suprasystemic right ventricular pressure 113/0 mmHg was measured, while the pressure in the left ventricle was 105/0 mmHg. The pressure in the aorta was 90/60 mmHg. Angiography confirmed SVAS with severe hypoplasia of both branches of the pulmonary artery.
During open heart surgery, the dimensions of the hypoplastic branches of the pulmonary artery were measured to be 5 mm for the right and 4 mm for the left one. The branches were consequently dilated with Hegar dilators to 7 mm for the right and 6 mm for the left branch. The arterial wall was found thickened to 1.5 mm. A Palmaz stent was inserted in the right pulmonary artery and expanded to 8–10 mm. After the operation, the gradient in the branches of the artery persisted. The histopathological analysis of the biopsy material from the trunk of the pulmonary artery showed normal intima, dysplastic changes in the media of the wall and partial fragmentation of elastic lamina, disorganization of their parallel organization with a tendency of being chaotic in certain parts. During catheterization 5 months later, the gradient in both branches was 90 mmHg. Angiography displayed dilated and hypertrophic ventricle. The left pulmonary artery was with severe hypoplasia and the stent in the right pulmonary artery was displaced peripherally. Although no heart failure and cyanosis have been detected, the diagnosis of severe hypoplasia and stenosis of both pulmonary arteries with thickened media led to the decision that the most effective solution for the stenosis would be balloon dilatation with cutting balloons.
Supravalvular aortic stenosis is a systemic elastin (ELN) arteriopathy that disproportionately affects the supravalvular aorta (reviewed in [Citation9]). ELN arteriopathy may be present in a nonsyndromic condition in patients present with normal intelligence and lack of dysmorphic features, or in syndromic conditions such as Williams–Beuren syndrome (WBS; OMIM 194 050). It is genetically heterogeneous and occurs as a consequence of haploinsufficiency of the ELN gene on chromosome 7q11.23, owing to either a microdeletion of the entire chromosomal region or ELN point mutations [Citation9].
Two pathological variants were found by DNA sequencing. The coordinates according to GRCh37/hg19 of the genetic variants are:
ELN: Chr7: g.73466253G>A, NM_000 501.2:c.890-1G>A (heterozygous);
SCN5A: Chr3:g.38674773C>A,NM_001099 404.1:c.26G>T, NP_001092 874.1:p.Gly9Val (heterozygous).
The variant ELN (c.890-1G>A) has not been described in medical literature so far and it disrupts a very conserved acceptor splicing site on the border of intron 15 and exon 16 (NM_000 501.2) in ELN. The mutation is expected to disrupt normal splicing, resulting in an abnormal protein product, so it is either pathogenic or likely pathogenic ().
Figure 1. Border between intron 15 and exon 16 of ELN.
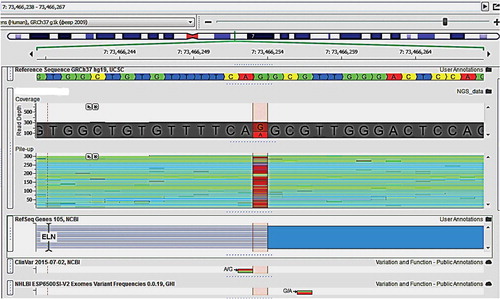
A similar acceptor splice-site mutation was described in a SVAS patient that causes both exon skipping and activation of a cryptic splice leading to a frameshift [Citation12]. The ELN gene has many small exons and elastin undergoes naturally occurring alternative splicing in vivo. Haploinsufficiency due to ELN mutations is usually the cause for SVAS, so it is possible that the mutant transcript is translated but the protein is not secreted or not incorporated into elastic fibres. Another possible mechanism was demonstrated in a study [Citation13] that described that the exclusion of exons because of mutations in the consensus splice junctions leads to aberrant tropoelastin that can lead to improper organization of the elastic fibres in the elastic matrix. This aberration was detected in SVAS and other disorders and suggests that these mutations are likely pathogenic, affecting the normal organization of elastic fibres in a dominant negative manner.
Many cases of pathological defects in ELN have been observed before. Seven novel mutations were detected in 31 familial and sporadic cases of nonsyndromic SVAS, including five frameshift mutations and two donor splice site mutations in intron 3 and 28 resulting in premature stop codons [Citation14]. In another study of a family suffering from SVAS, arterial stenosis, sudden death and intracranial aneurysm, a frameshift mutation was detected in exon 12 of ELN in the affected members [Citation15].
The second mutation in the same patient – a heterozygous substitution in SCN5A (p.Gly9Val) – has been reported before. It has been observed in patients with congenital long QT syndrome and patients with Romano–Ward syndrome characterized by irregular heartbeats due to longer recharge of the heart muscle, which can lead to cardiac arrest and death. Pathological variants in this gene have been reported in patients with different phenotypes as familial atrial fibrillation (OMIM 614 022 AD), Brugada syndrome 1 (OMIM 601 144 AD), Dilated cardiomyopathy 1E (OMIM 601 154 AD), nonprogressive heart block, progressive heart block type IA (OMIM 113 900 AD), Long QT syndrome-3 (OMIM 603 830 AD) and Sick sinus syndrome 1 (OMIM 608 567 AR) (http://omim.org/entry/600163). The three forms of primary electrical disorder, namely LQTS3, Brugada syndrome and cardiac conduction defects, have been associated with mutations in SCN5A [Citation16]. Most of the affected individuals were heterozygous carriers of a SCN5A mutation. A proband homozygous for a V1777M variant has been reported in a patient with major QT prolongation and 2:1 atrioventricular block [Citation17]. Compound heterozygosity in SCN5A was also described in a patient with a severe conduction defect in the proband and his deceased sister. A W156X mutation was inherited from the father and R225W, from the mother [Citation18]. In total, more than 150 mutations in SCN5A causing sodium channel dysfunction have been detected so far in patients with a number of arrhythmia syndromes [Citation19]. The combined heterozygosity of the mutations in ELN and SCN5A could explain the complex phenotype of our first patient.
The second patient was a 26-day-old baby first examined at the National Heart Hospital, Sofia, for evaluation of heart murmur. A rare form of asymmetrical left ventricle hypertrophy with mild left ventricular outflow obstruction (intra-ventricle gradient) was found. The patient was treated with calcium channel blocker (Diltiazem). Screening was conducted on the first-grade relatives. No similar symptoms were detected and cardiomyopathy was excluded in them. No heart anomalies were detected in the sister.
The patient was hospitalized at the age of 1 year and 9 months because of relapsing tachycardia continuing for a few hours without any notable provocation, second within two weeks. A sudden, short paroxysmal supraventricular tachycardia (250/min) occurred and was treated with Adenosine. Parameters measured indicated obstructive hypertrophic cardiomyopathy with hemodynamically significant gradient in the left ventricular outflow tract (LVOT).
Catheterization for inspecting the ventricle anatomy and function and invasive estimation of the gradient confirmed the diagnosis. The simultaneous measurement of the pressure in the femoral artery and the left ventricle showed dynamic gradient in the LVOT during sinus rhythm from 25 to 80 mmHg and a characteristic change in the pressure after extrasystole: drop in aortic pressure and rise in gradient to 100 mmHg (positive for Brockenbrough–Braunwald sign). Operative correction was suggested but not performed (myectomy by Morrow) with placement of electrocardiostimulator that will desynchronize the ventricle contraction in order to lessen the obstruction and protect from critical bradycardia under medical treatment. No serious complications in a period of more than two years were observed. The patient was treated with Beta blocker – 37.5 mg/day (3 × 12.5 mg). Upon the last regular exam of the patient, the electrocardiogram was without active ectopies of any kind and the LVOT gradient was fixed between 45–50 mmHg at rest. For now, no operation is intended.
During genetic analysis, two probably pathological variants were detected in the following genes:
CACNA1C: Chr 12:g.2675619A>G, NM_199 460.2:c.1540A>G, NP_955 630.2:p.Arg514Gly (heterozygous);
SCN5A: Chr3:g.38628928C>T,NM_001099 404.1:c.2399G>A, NP_001092 874.1:p.Arg800His (heterozygous).
Figure 2. Exon 12 of the CACNA1C gene in our second patient.
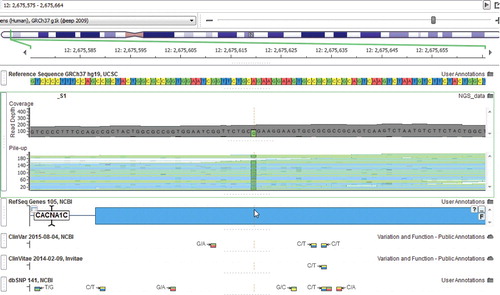
The effects of calcium channel mutations on cardiac arrhythmias were first reported in 2004 identifying a de novo mutation in CACNA1C as the cause for a multisystem disorder (Timothy Syndrome) [Citation20]. As many as 19 of the 55 known exons of the CACNA1C gene, which spans over 500 kb, are subject to alternative splicing and generate a diversity of expression products [Citation21]. A more recent study identified six CACNA1C mutations in seven probands out of 312 patients with Brugada Syndrome. The study also compared the clinical characteristics of patients with CACNA1C mutations to those with SCN5A mutations and found no differences in male predominance, age, symptoms or ECG characteristics [Citation22].
The second potentially pathological variant in our second patient was in the same gene as that in our first patient, SCN5A. The specific variant has been reported before and is associated with arrhythmogenic right ventricular cardiomyopathy. A study of three exons of SCN5A in a cohort of dilated and hypertrophic cardiomyopathy patients and controls identified many missense genetic variants classified as potentially damaging and disease-causing [Citation23]. Both parents of the second patient were healthy and showed no clinical features associated with HMC. Each of them was found to be a heterozygous carrier of only one of the two variants, which is in support of the digenic aetiology of the disease in their child. While most familial cardiomyopathies are monogenic disorders, our results indicate that the combination of two or more minor channel defects could be involved in the molecular pathophysiology of the disease.
A study of a cohort of 80 Australian unrelated HCM families identified dominant heterozygous mutations in 23 probands (29%) and compound or double heterozygote genotypes in four probands (5%). The results demonstrated that multiple gene mutations are associated with a more severe clinical phenotype than single heterozygous HCM patients, first being associated to increased left ventricular hypertrophy and a higher incidence of sudden cardiac death [Citation24]. Double heterozygosity was also associated with increased atrial arrhythmias, more severe LV thickening and an earlier age of onset in a family where the two mutations separately give a mild phenotype, but together result in a more severe phenotype [Citation25]. Another study also confirms that double-heterozygous patients had a higher extent of hypertrophy, earlier onset of disease and premature mortality [Citation26].
Identification of multiple mutations in an individual may change the necessary treatment and prophylaxis. The carrier status of more than one pathological channel mutation is a risk factor for sudden cardiac death, as well as a positive family history of sudden death and severe cardiac hypertrophy (>30 mm) [Citation24]. If only one disease mutation is detected in a double heterozygote, the omission of the other mutation could lead to possible false reassurance of the family members carrying the latter. Only the identification of a second mutation can give explanation for the more severe phenotypes, and failure to recognize double heterozygosity can lead to misdiagnosis. At least 16/38 (42%) cases with arrhythmogenic right ventricular cardiomyopathy (ARVC) were found to carry a combination of two mutations. Concomitant cause such as either a ‘second hit’ in the same gene (compound heterozygosity) or in a second gene (digenic heterozygosity) was shown to be required for the severe clinical phenotype [Citation27].
Conclusions
The diversity of phenotypes in cardiovascular diseases indicates that a wide range of genes needs to be tested, as complex inheritance of different variants can be the cause for more severe traits in some cases. The results from this study revealed that the application of high-throughput sequencing of large gene panels could reveal a more detailed genotype/phenotype relation in complex disorders such as some cardiovascular disorders.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethics standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Disclosure statement
The authors declare that they have no conflict of interests regarding the publication of this paper.
References
- Roger VL, Go AS, Lloyd-Jones DM, et al. Executive summary: heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation. 2012;125:188–197.
- Mendis S, Puska P, Norrving B. Global atlas on cardiovascular disease prevention and control. Geneva (Switzerland): World Health Organization; 2011. p. 3–18.
- Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the council on clinical cardiology, heart failure and transplantation committee. Circulation. 2006;113(14):1807–1816.
- Jarcho JA, McKenna W, Pare JA, et al. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med. 1989;321:1372–1378.
- Kathiresan S, Srivastava D. Genetics of human cardiovascular disease. Cell. 2012;148(6):1242–1257.
- Ashley EA, Hershberger RE, Caleshu C, et al. Genetics and cardiovascular disease: a policy statement from the American Heart Association. Circulation. 2012;126(1):142–157.
- Schäffer AA. Digenic inheritance in medical genetics. J Med Genet. 2013;50:641–652.
- Rodriguez-Revenga L, Badenas C, Carrió A, et al. Elastin mutation screening in a group of patients affected by vascular abnormalities. Pediatr Cardiol. 2005;26:827–831.
- Merla G, Brunetti-Pierri N, Piccolo P, et al. Supravalvular aortic stenosis: elastin arteriopathy. Circ Cadiovasc Genet. 2012;5(6):692–696.
- Gellens ME, George AL Jr, Chen LQ, et al. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci USA. 1992;89(2):554–558.
- Betzenhauser MJ, Pitt GP, Antzelevitch C. Calcium channel mutations in cardiac arrhythmia syndromes. Curr Mol Pharmacol. 2015;8(2):133–142.
- Urban Z, Michels VV, Thibodeau SN, et al. Supravalvular aortic stenosis: a splice site mutation within the elastin gene results in reduced expression of two aberrantly spliced transcripts. Hum Genet. 1999;104(2):135–142.
- Metcalfe K, Rucka AK, Smoot L, et al. Elastin: mutational spectrum in supravalvular aortic stenosis. Eur J Hum Genet. 2000;8(12):955–963.
- Micale, L, Turturo, MG, Fusco C, et al. Identification and characterization of seven novel mutations of elastin gene in a cohort of patients affected by supravalvular aortic stenosis. Eur J Hum Genet. 2010;18:317–323.
- Jelsig AM, Urban Z, Hucthagowder V, et al. Novel ELN mutation in a family with supravalvular aortic stenosis and intracranial aneurysm. Eur J Med Genet. 2017;60(2):110–113.
- Tan HL, Bink-Boelkens MT, Bezzina CR, et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature. 2001;409:1043–1047.
- Lupoglazoff JM, Cheav T, Baroudi G, et al. Homozygous SCN5A mutation in long-QT syndrome with functional two-to-one atrioventricular block. Circ Res. 2001;89(2):e16–e21.
- Bezzina CR, Rook MB, Groenewegen WA, et al. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ Res. 2003;92(2):159–168.
- Remme CA, Bezzina CR. Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc Ther. 2010;28(5):287–294.
- Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19–31.
- Tang ZZ, Liang MC, Lu S, et al. Transcript scanning reveals novel and extensive splice variations in human l-type voltage-gated calcium channel, Cav1.2 alpha1 subunit. J Biol Chem. 2004;279(43):44335–44343.
- Fukuyama M, Ohno S, Wang Q, et al. L-type calcium channel mutations in Japanese patients with inherited arrhythmias. Circ J. 2013;77(7):1799–1806.
- Priganc M, Zigova M, Boronova I, et al. Analysis of SCN5A gene variants in East Slovak patients with cardiomyopathy. J Clin Lab Anal. 2016 [cited 2017 Nov 26];31:e22037. DOI:10.1002/jcla.22037
- Ingles J, Doolan A, Chiu C, et al. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet. 2005 [cited 2017 Nov 26];42 (10): e59. DOI:10.1136/jmg.2005.033886
- van Rijsingen IA, Hermans-van Ast JF, Arens YH, et al. Hypertrophic cardiomyopathy family with double-heterozygous mutations; does disease severity suggest doubleheterozygosity?. Neth Heart J. 2009;17(12):458–463.
- Tsoutsman T, Bagnall RD, Semsarian C. Impact of multiple gene mutations in determining the severity of cardiomyopathy and heart failure. Clin Exp Pharmacol Physiol. 2008;35:1349–1357.
- Xu T, Yang Z, Vatta M, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2010;55(6):587–597.