
Abstract
Zhangzagu3, derived from Zhangzagu3fu and A2 (photoperiod- and thermo-sensitive genic male-sterile line) is well-known for its strong adaptability, good stability, disease, and lodging resistance. To evaluate the parental contributions to its elite traits, isobaric tags for relative and absolute quantitation (iTRAQ) and mass spectrometry (MS) were used to analyze the protein expression profile of these three lines. Enrichment analysis of differentially expressed proteins (DEPs) was performed through GO term and KEGG pathway. A total of 4015 proteins were detected, among which 504/390 DEPs between Zhangzagu3 and the female/male parent, respectively. The cluster analysis using Cluster 3.0 showed that the DEP expression profiles of Zhangzagu3 were closer to those of the male parent. GO functional classification divided the DEPs into three functional categories (cellular component, biological process and molecular function) and 71 subfamilies, among which three subfamilies had 100% difference between Zhangzagu3 and its parents. KEGG metabolic pathway exhibited significant enrichment of differentially expressed genes (DEGs) in Carbon metabolism (mainly Carbon fixation, Glycolysis and Gluconeogenesis), Photosynthesis and Oxidative phosphorylation. qRT-PCR verification confirmed the differential expression data and revealed that the expression level of 17 selected genes (such as putative receptor-like protein kinase, copper chaperone for superoxide dismutase etc.) was higher in Zhangzagu3 than that in its male parent, but significantly lower than that in A2. Thus, A2 can potentially optimize the agricultural qualities of Zhangzagu3. Taken together, we hope that this study provides valuable information for the further utilization and study on the molecular mechanisms of heterosis in hybrid millet.
| Abbreviations | ||
| iTRAQ | = | isobaric tags for relative and absolute quantitation |
| DEPs | = | differentially expressed proteins |
| DEGs | = | differentially expressed genes |
| PMSF | = | phenylmethanesulfonyl fluoride |
| HEPES | = | 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid |
| DTT | = | dithiothreitol |
| EDTA | = | ethylene diamine tetraacetic acid |
| GO | = | gene ontology |
| IAM | = | iodoacetamide |
| MS | = | mass spectrometry |
| KEGG | = | Kyoto Encyclopedia of Genes and Genomes |
| PVPP | = | polyvinylpolypyrrolidone |
| HPLC | = | high performance liquid chromatography |
| TCA | = | trichloroacetic acid |
| TEAB | = | tetraethylammonium bicarbonate |
| ACN | = | acetonitrile |
| HCD | = | higher collision energy dissociation |
| FDR | = | false discovery rate |
| SDS | = | sodium dodecyl sulphate |
| qRT-PCR | = | quantitative real-time polymerase chain reaction |
Introduction
As an important cereal crop and a model plant, foxtail millet (Setaria italica) has attracted much attention in China, India and some other Southeast Asian countries, where it is cultivated as a functional food owning to its high protein content, rich antioxidant components, dietary fiber and carotene [Citation1,Citation2]. This crop also exhibits advantage in having greater productivity, stronger fitness and shorter growing season than other crops under dry, high-temperature conditions [Citation3]. However, compared with the major crops (such as wheat, maize and rice), the yield of millet is much lower. To further improve its production and tolerance to adverse environment, the Zhangjiakou Academy of Agricultural Sciences bred a series of Zhangzagu millets using a two-line hybrid system [Citation4,Citation5]. The effective breeding of Zhangzagu improves the grain yield of millet and has made it become a high-yielding crop, which benefits to ensure food security and enrich deep-processing products. Among all hybrid millets, Zhangzagu3, derived from Zhangzagu3fu (male parent) and A2 (photoperiod- and thermo-sensitive genic male sterile line) exhibits significant hybrid vigour in its yield (26.67–46.67 kg/hm2) and resistance to drought and lodging [Citation4,Citation6,Citation7].
Hybrid vigour or heterosis refers to the phenomenon that hybrid descendants from crosses of inbred lines can display favourable phenotypes superior to their parents, such as higher yields, increased total biomass, extensive fitness and enhanced resistance to multiple stresses [Citation8]. Heterosis has long been used to improve agricultural production. Previous researches showed that the cotyledon size of Arabidopsis hybrids increased only a few days after sowing and accumulated higher vegetative biomass compared to their parents [Citation9–12]. Super Hybrid Rice, bred by the Father of Hybrid Rice, Longping Yuan, enjoys high popularity worldwide. Chaoyou 1000 for example, exhibits many elite properties, such as long photosynthesis duration, high seed-setting rate, and strong resistance to stresses and lodging. In the super high-yielding cultivation pattern, its total spikelet number reaches 750 million per hectare with sink potential for 1000 kg/667 m2 [Citation13]. Today, heterosis is applied as a key strategy to increase the grain yield in foxtail millet, and hybrid millet cultivars that have higher drought resistance and higher yields have been successfully developed [Citation14] through 30 years of research based on heterosis.
To gain insight into the mechanism of heterosis, a large number of new biotechnologies are widely used in this field. For example, the development of genome-wide expression technology, microarrays and high throughput sequencing allows researchers to acquire more information about gene expression profiles from hybrids and their parental lines. The differential expression of related genes on transcriptional level contributes to establishment of heterosis [Citation15,Citation16]. For instance, in the cotyledons of cabbage F1 hybrids, chloroplast-targeted genes were significantly up-regulated after 2 days of sowing, and the increased activity levels of these genes suggested that their differential transcription levels could be important in establishing early heterosis, becoming beneficial for later developmental vigour and yield advantage [Citation16].
To explore some of the mechanisms underlying the generation of heterosis at the protein level, proteomic molecular analysis has been introduced over many years [Citation17–19]. The abundance of a protein is not only regulated at a transcriptional level, but also at translational and post-translational levels [Citation20], so proteomics is a very powerful tool to analyze the functions of plant proteins [Citation21–23]. To identify a larger number of proteins and provide more reliable quantitative information, the iTRAQ (isobaric tags for relative and absolute quantitation) method has been introduced to execute quantitative comparison and analysis of protein expression profiles of multiple samples simultaneously [Citation24–26]. iTRAQ is a non-gel-based protein identification approach involving isotope labelling, and it has been proved to be one of the major quantification analysis tools in differential proteomic research [Citation27]. It has widely been applied to analyze the changes in protein expression under abiotic stresses [Citation20,Citation28,Citation29] or during growth and development [Citation30].
Zhangzagu3 millet has been verified to exhibit superior heterosis in grain yield and stress resistance, but there are few reports to explore the intrinsic mechanism. In this study, iTRAQ and mass spectrometry (MS) were employed to identify differentially expressed proteins (DEPs) between Zhangzagu3 and its parental lines; meanwhile, some bioinformatic methods (such as GO and KEGG) were used to analyze the functional enrichment of DEPs. This work shed light on the hybrid vigour of Setaria italica on the proteomic level and could provide a theoretical basis for the breeding of improved varieties in hybrid millet.
Materials and methods
Preparation of plant materials
The seeds of Zhangzugu3 and its parental lines were obtained from Zhangjiakou Academy of Agricultural Sciences, Hebei Province, China, and were grown in a 25 °C artificial incubator under a 12-h light/12-h dark cycle. Seedlings were harvested after 1 month, and the samples were washed with distilled water and stored at −80 °C for proteomic analysis.
Protein extraction
Total proteins were extracted from millet seedlings according to the following steps. The plant samples were ground in liquid nitrogen and PVPP (polyvinylpolypyrrolidone), and 10% TCA (trichloroacetic acid) in acetone (pre-cooled at −20 °C) was added to flush the ground material. After the samples were centrifuged (20,000g) for 30 min at 4 °C, the precipitate was washed and incubated with acetone for 30 min at −20 °C, and subsequently centrifuged (20,000g) at 4 °C for 30 min. Acetone washing was repeated until the precipitate turned white. Thereafter, protein extraction reagent, was added, which is composed of 1 mmol/L PMSF (phenylmethanesulphonyl fluoride), 30 mmol/L HEPES (4-(2-hydroxyethyl)-1-piperazine ethanesulphonic acid), 8 mol/L urea, 2 mmol/L EDTA (ethylene diamine tetraacetic acid) and 10 mmol/L DTT (dithiothreitol). And the pellets were dissolved by ultrasound (pulse on 2 s, off 3 s, power 180 W) for 5 min. Then, after centrifugation at 20,000g and 4 °C for 30 min, the pellets were discarded and the protein fraction in the supernatant was used in the subsequent steps. Protein concentration was determined using Bradford assay Kit (GE Healthcare, Pittsburgh, PA, USA) following the manufacturer’s instructions.
Protein digestion and iTRAQ labelling
The iTRAQ analysis was performed by Beijing Genomics Institute (BGI, Shenzhen, China). For each protein sample, 100 μg of protein was incubated with 10 mmol/L DTT at 56 °C for 1 h, followed by 1 h of incubation with IAM (iodoacetamide) in darkness, 3 h of precipitation in acetone at −20 °C, and centrifugation at 20,000g and 4 °C for 30 min in given order. The precipitate was re-suspended and ultrasonicated in pre-chilled 50% TEAB (tetraethylammonium bicarbonate) buffer with 0.1% SDS (sodium dodecyl sulphate). The proteins in TEAB were digested with trypsin at 37 °C for 24 h, and the digested samples were dried in a centrifugal vacuum concentrator. The protein lyophilized powder was dissolved in 30 μL of 50% TEAB (Sigma, St. Louis, MO) together with 70 μL of isopropanol and the samples were labeled with the iTRAQ reagents. In brief, all samples were respectively labelled with iTRAQ tags 117, 118 and 119.
High performance liquid chromatography (HPLC)
The labelled samples were fractionated using an HPLC system. The peptides were eluted using Buffer A (10 mmol/L KH2PO4, 25% acetonitrile and acidified to a pH value of 3.0 with H3PO4) and Buffer B (10 mmol/L KH2PO4, 2 M KCl, 25% acetonitrile and acidified to a pH value of 3.0 with H3PO4). The gradient elution of the chromatographic column was conducted as follows: 0–35 min 100% Buffer A; 36 min 5% Buffer B; 56 min 30% Buffer B; 61 min 50% Buffer B, 66 min 50% Buffer B; 71 min 100% Buffer B. The flow rate was set at 1 mL/min, and fractions were collected every minute after sample injection for 31 min. Based on the chromatogram, 42 collected elution ingredients were merged into 18 samples, which were desalted using C18 solid-phase extraction cartridge and vacuum dried. C-18 column was activated by 1 mL methanol at a speed of 2–3 drops/s, and then was balanced by 5% ACN (acetonitrile) at a speed of 1 drop/s.
LC-MS/MS and data analysis
All freeze-dried samples were dissolved in 0.1% methane acid, loaded onto the C18-reversed phase column (75 μm × 2 cm, 3 μm, 100 Ǻ, Thermo Scientific) and separated with a reversed phase C18 column (100 mm ×75 μm, 300 Å, 5 μm) mounted on the nLC system. Peptides were eluted with a gradient of 5–80% (v/v) acetonitrile in 0.1% formic acid at a 400 nL/min flow rate in 45 min. Subsequently, 18 samples were directly injected Q-E MS, respectively. All parameters of MS analysis are described in . For reliable statistical consistency, three biological replicates were set for better coverage of the target proteome. All MS raw files were processed by Mascot 2.2 and Proteome Discoverer 1.3 (Thermo Fisher Scientific, SanJose, CA, USA) to identify proteins. All acquired MS/MS spectra were automatically searched in Setaria_uni_4554 database. With at least two unique peptides and with a value of FDR <0.01, a unique protein was qualified for further quantification analysis. FDR correction was adopted with a threshold of 0.01 to reduce the false identification of peptide, a Mascot probability of identification and quantification of protein. Protein identification requests detection of one unique peptide at least.
Table 1. Parameters of MS analysis.
Cluster analysis
Cluster analysis of the proteins identified in Zhanggu3fu, Zhangzagu3 and A2 was carried out using Cluster 3.0 software (Michael Eisen, Stanford University), and these proteins were clustered depending on their expression level.
Bioinformatics
In order to determine the function of DEPs in the metabolism pathways and their destined biological functions, we used online software products for the analysis of GO terms (http://www.geneontology.org/) and KEGG pathway (http://www.genome.jp/kegg/).
RNA extraction and qRT-PCR analysis
The plant samples were ground in liquid nitrogen and TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and total RNA was extracted according to the manufacturer’s instructions. Then, cDNA was synthesized in the following reaction system: 10 μL 2 × RT buffer, 1 μL 6 N Random primer (100 pmol/μL), 1 μL RT-mix, 5 μL template (RNA), 3 μL DEPC (diethy pyrocarbonate) water. The reaction program was set as follows: 25 °C for 10 min, 42 °C for 50 min, 85 °C for 5 min, and the reaction was performed using PCR amplifier (ABI ProFlex). The primers for qRT-PCR analysis were designed using Primer 5 software. To ensure all primers were specific to their targeted gene, the primer sequences were blasted in the NCBI database (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). Details about the selected genes and their primers are shown in Supplementary material . The 18 S rDNA gene was used as internal reference for normalization. The qRT-PCR reaction system was prepared with the following reagents: 2 × PCR buffer, 25 μL; 25 pmol/μL forward primer, 1 μL; 25 pmol/μL reverse primer, 1 μL. SYBR® Green I, 0.5 μL; template (cDNA), 2 μL; DEPC water, 20.5 μL. The PCR parameters were: 94 °C for 4 min, followed by 35 cycles of 94 °C for 20 s, 60 °C for 30 s and 72 °C for 30 s. The signal was detected at 72 °C.
Results and discussion
Primary data analysis and protein identification
Proteins extracted from seedlings of Zhangzugu3 and its parental lines were analyzed based on the iTRAQ experiment, and a total of 345688 spectra were generated. Mascot revealed 76777 spectra matched to the known spectra, 23,063 peptides matched to the known peptides, 18,102 unique peptides and 4015 proteins (, Supplementary Material ). The distribution of peptide length, peptide number, mass and sequence coverage of proteins are shown in . About 57% of the proteins contained at least 4 peptides. The molecular weight of most proteins was about 20–50 kDa. Protein sequences coverage with 40–100%, 35–40%, 30–35%, 25–30%, 20–25%, 15–20%, 10–15%, 5–10% and under 5% accounted for 13.8%, 4.3%, 5.4%, 6.4%, 7.8%, 10.3%, 13.9/5, 19.0% and 19.1%. (e.g. 40–100% means more or equal to 40%, and less than 100%).
Figure 1. Spectra, peptide and proteins identified based on iTRAQ proteomics by searching against the Setaria_ uni_4554 database.
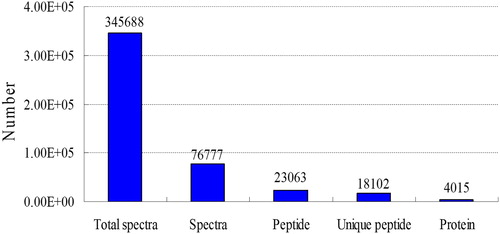
Figure 2. Distribution of peptide length (A), number of peptides that are matched to proteins using MASCOT (B), protein mass distribution (C), and distribution of protein sequence coverage (D) identified by iTRAQ proteomics.
Table 2. Number of differentially expressed proteins in seven categories.
DEPs detected by iTRAQ
As a powerful means in proteomics research, iTRAQ can identify a greater number of proteins and provide more reliable information than two-dimensional electrophoresis (2-DE) [Citation30]. In this present study, a total of 4015 proteins were detected based on the iTRAQ-mediated proteomic analysis of 1-month-old plants from Zhangzagu3 and its parents (Supplementary Material ). When the expression level of one protein exhibited a difference (a > 2-fold or <0.5-fold) compared to the control line, it was considered to be differentially expressed. There were hundreds of DEPs with pairwise comparison among these three lines (, Supplementary Material ). When Zhangzagu3 was taken as the control line, A2 (female parent) displayed 504 DEPs, among which 229 presented higher expression and 275 showed lower expression; whereas the male line had 390 DEPs: 156 displayed higher expression and 234 showed lower expression. And there were 724 DEPs between Zhangzagu3fu and A2, including 302 ones with higher expression and 422 ones with lower expression. Thus, the similarity between Zhangzagu3 and its male parent was higher than that between Zhangzagu3 and its female parent or between the two parental lines themselves.
Figure 3. Venn diagram of differentially expressed proteins in Zhangzagu3 and its parents.
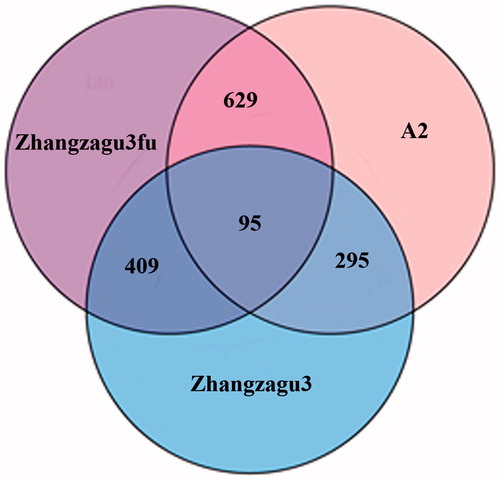
Table 3. Gene Ontology (GO) functional annotation of DEPs between Zhangzagu3 and its parents (P < 0.05, the top 5).
Table 4. Up-/down-regulated parental genes involved in the main biological processes.
According to their specificity to these three lines, the DEPs were classified into 7 categories (). Category 1 includes 85 proteins that were differentially expressed between Zhangzagu3 and its female parent (A2). In Category 2, 140 DEPs corresponded to Zhangzagu3 and its male parent. Category 3 contains 214 proteins that were differentially expressed between both parental lines. Category 4 was composed of 178 DEPs between the two parental lines and between Zhangzagu3 and A2. Category 5 presented 237 proteins which were differentially expressed between two parental lines and between Zhangzagu3 and Zhangzagu3fu. Category 6 exhibited 32 proteins expressed differentially between Zhangzagu3 and both parental lines. Lastly, Category 7 consisted of 95 proteins specific to all three lines.
Protein expression pattern analysis
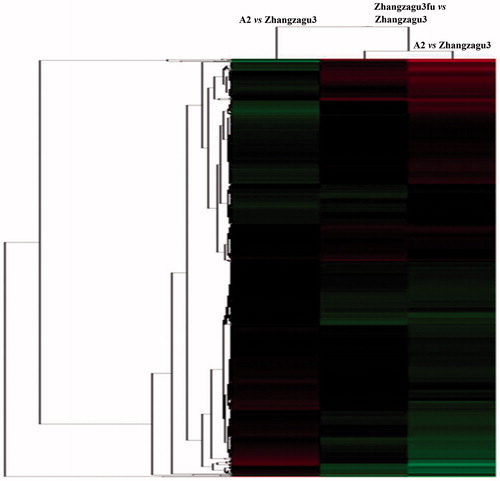
The examination of DEPs between Zhangzagu3 and its parental lines leads to a good understanding of the relevance of protein expression profiles and agronomic traits. A cluster analysis of the proteins identified in these three cultivars was performed using Cluster 3.0 software, and the proteins were clustered depending on their expression level. As shown in , the expression patterns of DEPs between Zhangzagu3 and A2 shared a slightly lower similarity than those of Zhangzagu3 and Zhangzagu3fu, and the similarity of Zhangzagu3 to both parents was higher than that between its two parents. Therefore, the DEP expression profiles of Zhangzagu3 were closer to those of the male parent. Therefore, it was estimated that the agronomic effects exerted on Zhangzagu3 by the male parent were greater than those by the female parent.
Figure 4. Cluster map comparing the protein expression patterns of Zhangzagu3, Zhangzagu3fu and A2. Note: Red indicates higher expression, green indicates lower expression, and black indicates the same expression levels in the two strains.
GO functional classification
To analyze the function of DEPs between the hybrid cultivar and its parents, GO functional classification was performed using the GO online tool (http://www.geneontology.org/). The GO term enrichment analysis revealed that all DEPs with significant difference (P < 0.05) are divided into three groups and 71 subfamilies (Supplementary Material ). The 1st group based on the cellular composition consists of 27 functional protein subfamilies; the 2nd group involved in the biological processes had 31 subfamilies, and the proteins in the 3rd group mainly participated in molecular functions. As shown in , DEPs functioning in negative regulation of reductive pentose-phosphate cycle, regulation of photosynthesis of dark reaction and regulation of reductive pentose-phosphate cycle, were considered to represent 100% difference. And compared to other groups, there was least differential expression in the ‘Cellular component’ group.
Figure 5. Proteins differentially expressed between Zhangzagu3 and its parents. Note: GO analysis results.
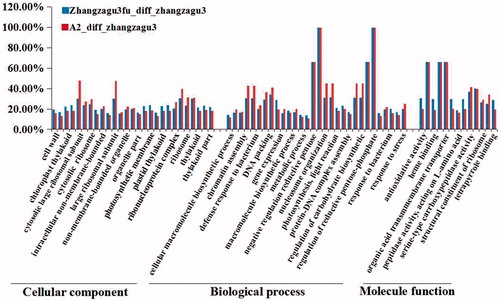
Table 5. DEP-enriched metabolic pathways (P < 0.05, the top ten).
In order to reveal the significant enrichment of DEPs, the data in Supplementary Material was organized, and the top five subfamilies in the aforementioned three groups were identified (). For example, the 2nd group, compared to others, was more enriched with DEPs associated with the biological processes of response to stress, gene expression, generation of precursor metabolites and energy, Photosynthesis and DNA packaging. Thus, these DEPs were found to be mainly enriched in the biological processes of Response to stresses and energy metabolism, which may be good for improvement of the hybrid vigour of Zhangzagu3. It is well known that great tolerance to stresses and powerful photosynthesis efficiency benefit plant growth and production. For example, previous researches reported that 90% dry matter of millet was derived from its photosynthetic product, and uttermost improvement of light use efficiency will exert an important influence on the increase of grain yield [Citation31–33]. The hybrid vigour of crops is related to photosynthesis and it may allow for a new method of selecting new species with high photosynthesis rate [Citation34]. For instance, in early Arabidopsis development (at 3 d after sowing) the increase in the expression activity of genes involved in photosynthesis leads to increases in cell size and number, which suggests a central role for enhanced photosynthesis in the production of the heterotic biomass [Citation10].
After the expression profiles of DEPs involved in Response to stress and Photosynthesis were analyzed (), more than 50% DEPs were revealed to represent hyper-parent expression. It was deemed that enhanced expression of positive effect genes and suppressed expression of negative effect genes both contributed to the heterosis effect in the hybrid.
KEGG pathway enrichment of DEPs
To study the changes of metabolic pathways between the cross combination ‘A2 and Zhangzagu3fu’, KEGG pathway analysis was performed with the DEPs, and 68 different metabolic pathways were summarized (Supplementary Material Table S6). As shown in , the enrichment of differentially expressed genes (DEGs) was significant in Carbon metabolism (mainly including Carbon fixation, Glycolysis and Gluconeogenesis), Photosynthesis and Oxidative phosphorylation. The categories of Metabolism of Amino acid, Amino sugar, Starch and sucrose were also enriched with dozens of DEGs. In addition, consistent with GO classification, a number of DEPs were involved in the metabolic pathways corresponding to stress responses, such as Glutathione metabolism. These pathways are known to have some relationship with crop growth and yield. Thus, DEP enrichment in these metabolic pathways may be in favour of the formation of hybrid vigour.
qRT-PCR verification
To further confirm the DEPs between Zhangzagu3 and its two parents, 17 differentially expressed genes were chosen from three groups mentioned above, and qRT-PCR was carried out. In the first group, eight of the chosen genes were involved in the catalysis of phosphorylation. Two genes were selected from the second group, which is involved in biological functions. In the third group, seven genes were chosen and they have no common function; namely, they were respectively predicted to encode G-type lectin S-receptor-like serine/threonine-protein kinase, tonoplast dicarboxylate transporter, potassium transporter, copper chaperone for superoxide dismutase, two-on-two hemoglobin, heat stress transcription factor, bark storage protein (, Supplementary Material ). Of all the genes, two ones exhibited extremely high expression in A2. One encodes a receptor-like kinase protein (LOC101768960), and the other one, the copper chaperone for superoxide dismutase in chloroplast (LOC101781151). Based on the InterPro database, the N-terminal of receptor-like serine/threonine-protein kinase has a glycine-rich stretch of residues involved in ATP binding, which has close relationship with energy metabolism [Citation35,Citation36]; and the copper chaperone for superoxide dismutase has a heavy-metal-associated domain (HMA), composed of approximately 30 amino acid residues that play a key role in transporting or detoxifying heavy metals like copper chaperones [Citation37,Citation38]. This can enhance plant tolerance in response to long-term toxicity of heavy metals. Thus, if the energy utilization and resistance to stresses of Zhangzagu3 were further enhanced, its agronomic traits would obtain further improvement.
Figure 6. qRT-PCR verification of some differentially expressed proteins. Note: The proteins corresponding to the IDs can be seen in Supplementary Information Table S1. Data are mean values with standard deviation (means ± SD) of three separate experiments.
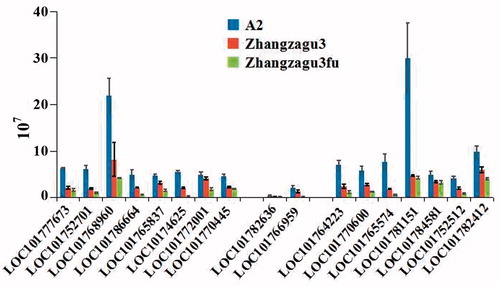
These 17 selected genes are mainly involved in phosphorylation regulation and stress response (), and it was aforementioned that these two biological functions have close relationship with heterosis formation. As shown in , the expression level of these 17 genes was lower in Zhangzagu3 than in A2, but higher than that in the male parent. Therefore, based on the male parent, Zhangzagu3fu, the male-sterile line A2 as the female parent can improve the expression ability of genes related to phosphorylation regulation and stress response in its hybrid progeny, Zhangzagu3, thus contributing to the elite agriculture traits of Zhangzagu3.
Conclusions
In this study, we used the iTRAQ technique to analyze the protein expression profiles of Zhangzagu3 and its parental lines. The results showed that 4015 proteins in all were detected. The cluster analysis revealed that the DEP expression profiles of Zhangzagu3 were closer to those of the male parent. The function analysis of GO term and KEGG pathway showed that DEPs were significantly enriched in the categories/groups of synthesis of organic compounds, energy production and stress response, which was hypothesized to result in heterosis. We found that the female parent (A2) can improve the physiological and agronomic characteristics of its progeny on the basis of Zhangzafu3fu. We hope our proteomics analysis of differential expression in hybrid millet and its parents can provide valuable information for effective application of heterosis in breeding hybrid millet.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Kalinova J, Moudry J. Content and quality of protein in proso millet (Panicum miliaceum L.) varieties. Plant Food Human Nutr. 2006;61:45–49.
- Xu YB, Li JY, Wan JM. Agriculture and crop science in China: innovation and sustainability. Crop J. 2017;5:95–99.
- Diao XM. Production and genetic improvement of minor cereals in China. Crop J. 2017;5:103–114.
- Lu HB, Gong XC, Qiao YM, et al. A study on dry matter accumulation and light characteristics of hybrid foxtail millet. Crops. 2014;35:121–124.
- Wang XM, Song GL, Feng XL, et al. Research of high yield propagation technique for photo-thermo-sensitive genic male sterility of foxtail millet. J Heibei Agric Sci. 2016;20:5–9.
- Fan GY, Zhao ZH, Yuan JC, et al. Study the relationship between anatomical structure and biology characters of hybrid millet Zhangzagu 3 hao. Acta Agric Boreali-Sinica. 2014;29:114–119.
- Yang JY. High-yield cultivation techniques of zhangzagu 3 hao. Seed Sci Technol. 2010;28:43–44.
- Zhang L, Peng YG, Wei XL, et al. Small RNAs as important regulators for the hybrid vigour of super-hybrid rice. J Exp Bot. 2014;65:5989–6002.
- Groszmann M, Greaves IK, Albertyn ZI, et al. Changes in 24-nt siRNA levels in Arabidopsis hybrids suggest an epigenetic contribution to hybrid vigor. Proc Natl Acad Sci USA. 2011;108:2617–2622.
- Fujimoto R, Taylor JM, Shirasawa S, et al. Heterosis of Arabidopsis hybrids between C24 and Col is associated with increased photosynthesis capacity. Proc Natl Acad Sci USA. 2012;109:7109–7114.
- Meyer RC, Witucka-Wall H, Becher M, et al. Heterosis manifestation during early Arabidopsis seedling development is characterized by intermediate gene expression and enhanced metabolic activity in the hybrids. Plant J. 2012;71:669–683.
- Groszmann M, Gonzalez-Bayon R, Greaves IK, et al. Intraspecific Arabidopsis hybrids show different patterns of heterosis despite the close relatedness of the parental genomes. Plant Physiol. 2014;166:265–280.
- Wu ZH, Sun QH, Dong YX, et al. Performance and cultivation techniques of a promising super hybrid rice combination Chaoyou 1000 in high-latitude Area. Agric Sci Technol. 2017;18:1012–1015.
- Liu ZL, Bai GH, Zhang DD, et al. Heterotic classes and utilization patterns in Chinese foxtail millet [Setaria italica (L.) P. Beauv]. Agric Sci. 2014;5:1392–1406.
- Li L, He X, Zhang H, et al. Genome-wide mapping reveals a combination of different genetic effects causing the genetic basis of heterosis in two elite rice hybrids. J Genet. 2015;94:261–270.
- Saeki N, Kawanabe T, Hua Y, et al. Molecular and cellular characteristics of hybrid vigour in a commercial hybrid of Chinese cabbage. BMC Plant Biol. 2016;16:45–59.
- Qin J, Gu F, Liu D, et al. Proteomic analysis of elite soybean Jidou17 and its parents using iTRAQ-based quantitative approaches. Proteome Sci. 2013;11:12. DOI:10.1186/1477-5956-11-12
- Baranwal VK, Mikkilineni V, Zehr UB, et al. Heterosis: emerging ideas about hybrid vigour. J Exp Bot. 2012;63:6309–6314.
- Schnable PS, Springer NM. Progress toward understanding heterosis in crop plants. Annu Rev Plant Biol. 2013;64:71–88.
- Jiang HX, Yang LT, Qi YP, et al. Root iTRAQ protein profile analysis of two Citrus species differing in aluminum-tolerance in response to long-term aluminum-toxicity. BMC Genomics. 2015;16:949–965.
- Dong K, Ge P, Ma C, et al. Albumin and globulin dynamics during grain development of elite Chinese wheat cultivar Xiaoyan 6. J Cereal Sci. 2012;56:615–622.
- Lesage VS, Merlino M, Chambon C, et al. Proteomes of hard and soft near-isogenic wheat lines reveal that kernel hardness is related to the amplification of a stress response during endosperm development. J Exp Bot. 2012;63:1001–1011.
- Ge P, Ma C, Wang S, et al. Comparative proteomic analysis of grain development in two spring wheat varieties under drought stress. Anal Bioanal Chem. 2012;402:1297–1313.
- Ghosh D, Li Z, Tan XF, et al. iTRAQ based quantitative proteomics approach validated the role of calcyclin binding protein (CacyBP) in promoting colorectal cancer metastasis. Mol Cell Proteomics. 2013;12:1865–1880.
- Huang X, Zeng Y, Xing X, et al. Quantitative proteomics analysis of early recurrence/metastasis of huge hepatocellular carcinoma following radical resection. Proteome Sci. 2014;12:22–35.
- Kambiranda D, Katam R, Basha SM, et al. iTRAQ-based quantitative proteomics of developing and ripening muscadine grape berry. J Proteome Res. 2014;13:555–569.
- Wang LQ, Dai Y. iTRAQ labeling and biomarker discovery in comparative proteomic studies. Chem Life. 2015;30:135–140.
- Liu JY, Men JL, Chang MC, et al. iTRAQ-based quantitative proteome revealed metabolic changes of Flammulina velutipes mycelia in response to cold stress. J Proteomics. 2017;156:75–84.
- Hu XL, Li NN, Wu LJ, et al. Quantitative iTRAQ-based proteomic analysis of phosphoproteins and ABA-regulated phosphoproteins in maize leaves under osmotic stress. Sci Rep. 2015;5:15626–15651.
- Ma CY, Zhou JW, Chen GX, et al. iTRAQ-based quantitative proteome and phosphoprotein characterization reveals the central metabolism changes involved in wheat grain development. BMC Genomics. 2014;15:1029–1049.
- Zhang ZW, Qu JB, Xu GF, et al. Maize and foxtail millet as substantial sources of dietary lead intake. Sci Total Environ. 1997;208:81–88.
- Xiao J, Yang T, Yang Z, et al. Analysis on gene expression in rice under endophytes alcohol extraction treatment with gene microarray technique. Biotechnol Bull. 2013;1:89–95.
- Yang YJ, Zhao HM, Shao QL, et al. Comparison of photosynthetic and chlorophll fluoresence between conventional and hybird foxtail millet. Crops. 2015;3:146–149.
- Zeng B, Xu XM, Zhou SX, et al. Effects of temperature and light on photosynthetic heterosis of an upland cotton hybrid cultivar. Crop Sci. 2012;52:282–291.
- Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596.
- Knighton DR, Zheng JH, Ten-Eyck LF, et al. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–414.
- Banci L, Bertini I, Ciofi-Baffoni S, et al. Solution structure of the yeast copper transporter domain Ccc2a in the apo and Cu (I)-loaded states. J Biol Chem. 2001;276:8415–8426.
- Xiao Z, Loughlin F, George GN, et al. C-terminal domain of the membrane copper transporter Ctr1 from Saccharomyces cerevisiae binds four Cu(I) ions as a cuprous-thiolate polynuclear cluster: sub-femtomolar Cu(I) affinity of three proteins involved in copper trafficking. J Am Chem Soc. 2004;126:3081–3090.