
Abstract
Picea neoveitchii Mast. is a rare and endangered evergreen coniferous tree species in China. P. neoveitchii grows within a specific ecological and geographical range. Here, we performed transcriptome sequencing of P. neoveitchii using the Illumina high-throughput deep sequencing technology with the following aims: (i) to perform de novo transcriptome assembly, (ii) to perform transcriptome assembly with functional annotation, (iii) to study the candidate genes involved in the biosynthesis of cellulose and lignin, (iv) to identify and analyse the transcription factors, and (v) to develop potential expressed sequence tag–simple sequence repeat (EST-SSR) markers for future population genomic and genome-wide association studies in P. neoveitchii. This is, to the best of our knowledge, the first study to provide a comprehensive transcriptome of P. neoveitchii. The transcriptome data contained 12.97 gigabase pairs (Gbp) for a total of 120,951 different transcripts belonging to 66,022 unigenes. In addition, we classified enzymes involved in the biosynthesis of cellulose and lignin and identified and analysed the transcription factors involved in stress responses, growth and development. Based on the larger than 1 kb unigenes, 1,814 EST-SSR markers were shown. Our results demonstrated that the transcriptome analysis is a valuable technology for non-model species with large genomes and this dataset will be an essential resource for future genetic and genomic research in P. neoveitchii.
Introduction
Picea neoveitchii Mast. is an evergreen coniferous tree that belongs to the genus Picea in the Pinaceae family. This species is both rare and an endangered species that is native to China. It grows within an ecological and geographical range that is severely threatened by climate change, forest fires and land-use changes [Citation1,Citation2]. This species is important to various disciplines, including botany, geography and climatology. When compared with other conifers, this conifer has a straight and vertical trunk, and its fibre and wood are of a very high quality. Furthermore, whereas its cones contain compounds that can be used for the treatment of cough and phlegm, its leaves contain compounds that can inhibit fungal damage to plants [Citation3,Citation4]. So far, research into the protection and utilisation of Picea neoveitchii has focused on its wood and leaf extracts, and its genomics and transcriptomics have not been explored.
Recent studies have shown that RNA-Seq is a very effective and powerful technique for gathering extensive transcriptome information in many plants [Citation5–8]. When compared with other hardwood species [Citation9–15], conifers tend to have larger and highly repetitive genomes [Citation16–21]. Sequencing the P. neoveitchii genome remains expensive, even when using high-throughput Illumina sequencing technology. However, better transcriptome sequencing results can provide information about the gene expression regulation and the amino acid sequences encoded by certain genes.
This study focused particularly on the classification of the assembled unigenes involved in the biosynthesis of cellulose and lignin. Cellulose is an important part of the plant cell wall and is considered to be the most abundant polysaccharide material in nature, with an average annual production and decomposition of about 180 billion tons [Citation22,Citation23]. Lignin is also an important part of the plant cell wall; it is a highly branched polymer composed of phenylpropanoids and represents about 30% of the organic carbon in the biosphere [Citation24,Citation25]. Using this new comprehensive transcriptome dataset, we can also further study some important functional genes and determine some molecular markers for transcriptional genomics, such as ones for stress responses, growth and development. These data will be an essential resource for future genetic and genomic research.
In this study, we obtained the P. neoveitchii comprehensive transcriptome sequence by Illumina RNA-Seq technology. The transcriptome is publically available in the NCBI (National Center for Biotechnology Information) database, and we believe that it will help future researchers to perform breeding, gene expression and population genomic analyses of this coniferous tree species. At the same time, the study will contribute to the study of the genetic evolution of the entire gymnosperms.
Materials and methods
Plant materials and RNA extraction
Root, leaf and stem samples from a single individual P. neoveitchii tree were collected. The samples were snap frozen in liquid nitrogen and stored for subsequent experiments. We used the Plant RNA Kit (Aidlab-Biotech, China) to extract total mRNA. A spectrophotometer and an Agilent 2100 Bioanalyzer were used for measuring the quality and quantity of purified mRNA. Subsequently, we combined the same amount of high quality mRNA from each tissue to produce a pooled mRNA sample for the mRNA-seq library.
Library construction for illumina sequencing, data analysis and assembly
The Illumina sequencing library was constructed by the Illumina’s TruSeq RNA Sample Preparation Kit. The quality of the library was assessed by the Agilent Bioanalyzer 2100 system. The fragments (300 bp ± 25 bp) were purified and amplified as previously reported in detail [Citation5], and then the library was sequenced using the Illumina HiSeq platform.
In order to obtain high-quality clean data, we removed all adapter sequences and low-quality sequences from the raw data. High quality data reads were de novo assembled using the Trinity program (http://trinityrnaseq.sourceforge.net/) [Citation26]. Our optimised k-mer length was 25. The longest assembled transcripts were defined as unigenes. The coding area was predicted by the EMBOSS Getorf Software (http://emboss.bioinformatics.nl/cgi-bin/emboss/getorf) [Citation27]. The transcriptome is publically available in the NCBI (National Center for Biotechnology Information) database.
Functional annotation
Annotating the unigenes was done based on the following six databases: NR, NT, SWISS-PROT, GO, COG and KEGG. We selected the best alignment from the matches with an E-value ≤ 10−5. Based on the highest score BLAST comparison, we gave a gene name for each assembled unigene sequence. We assigned GO to the assembled unigene sequence using the “Blast2GO” [Citation28]. The Kyoto Encyclopedia of Genes and Genomes pathways were assigned to the assembled sequences using the online KAAS (http://www.genome.jp/kegg/kaas/).
EST-SSR detection
We used the MISA software to perform EST-SSR detection in the assembled 12,845 unigenes (≥1kb) (http://pgrc.ipk-gatersleben.de/misa/) by adjusting the parameters to identify perfect Di-, Tri-, Tetra-, Penta- and Hexa-nucleotide motifs with a minimum of six, five, four, four and four repeats respectively as previously described [Citation5].
Results and discussion
RNA-Seq and transcriptome assembly
After removing all adapter sequences and low-quality sequences from the raw data, about 64,221,704 reads (12.97 Gbp) with 91.44% Q20 bases were selected (see Supplemental Table S1). High quality Illumina sequencing reads were submitted to the NCBI Short Read Archive (accession number: SRR2132464).
We used the Trinity software program to de novo assemble the high-quality sequencing data [Citation19], which produced 120,951 transcripts with an N50 length of 1,467 bp and a mean length of 871 bp. The length distribution of the transcripts is shown in Supplemental Figure S1A and Table S2. Then the assembled transcripts were further analysed for clustering and assembly. This produced 66,022 unigenes with an N50 length of 1,194 bp and a mean length of 668 bp ( and Supplemental Figure S1B). These data will be used for subsequent annotation analysis.
Table 1. Summary of P. neoveitchii unigenes.
Functional annotation and classification
The unigenes functional annotation is shown in . We used BLASTX for similarity analysis and compared against the NR database. Of these unigenes, 41,011 (62.12%) unigenes had a significant match, whereas approximately 40% of the unigenes did not show a significant match. Based on the previous studies, it depicts that the determined sequences of unigenes did not significantly match about 35%-50% of the sequences. Among the many plant protein sequences, the assembled unigenes of P. neoveitchii had the highest number of hits against Picea sitchensis at 12,205 hits (29.76%), followed by Vitis vinifera at 2,896 hits (7.06%), Glycine max at 1,784 hits (4.35%), Theobroma cacao at 1,295 hits (3.16%), Ricinus communis at 1,227 hits (2.99%), and Populus trichocarpa at 1,086 hits (2.65%) (). The high similarity of Picea neoveitchii unigenes and Picea sitchensis genes indicates that we can use the Picea sitchensis transcriptome and genome as a reference for further study.
Figure 1. Species distribution of the top BLAST hits in the NR database.
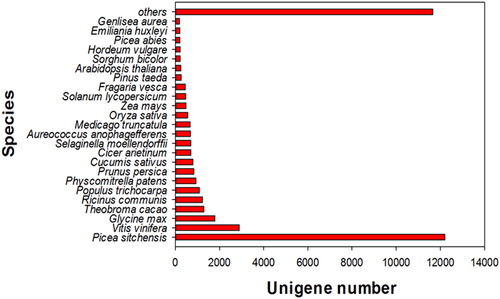
Table 2. Functional annotation of the P. neoveitchii transcriptome.
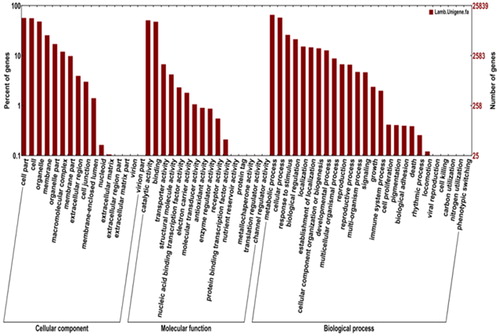
The SWISS-PROT database annotation results contain experimental results, computational features and scientific conclusions. Of the 66,022 assembled unigenes, 30,924 (46.84%) were similar to accessions in SWISS-PROT. Using the GO database for enrichment analysis, the unigenes could be classified into three separate groups. The most GO terms were assigned to biological process 67,447 (48.40%); molecular function had 38, 975 (27.97%) terms assigned, and cellular component had 32,927 (23.63%) terms assigned ().
Figure 2. Gene Ontology (GO) classification of assembled unigenes in P. neoveitchii.
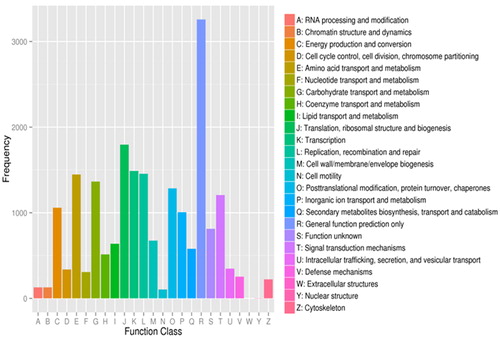
In addition, the assembled unigenes were searched against the COG database. A total of 15,737 unigenes were similar to accessions in the COG database (). The highest term was that for the general function prediction cluster (3,257, 15.95%), followed by translation, ribosomal structure and biogenesis (1,795, 8.79%); transcription (1,488, 7.29%); replication, recombination and repair (1,455, 7.13%); amino acid transport and metabolism (1,477, 7.09%); carbohydrate transport and metabolism (1,365, 6.69%); posttranslational modification, protein turnover and chaperones (1,285, 6.29%); signal transduction mechanisms (1,207, 5.91%); and energy production and conversion (1059, 5.19%). However, only 3 assembled unigenes were annotated to the nuclear structural components, and no unigene was annotated to the extracellular structures. The KEGG database was used to analyse the active biological metabolic pathways. Annotated analysis found that 12,302 assembled unigenes were assigned to 206 biological pathways. Among them, the more highly represented KEGG pathways were the ribosome pathway (ko03011,922 unigenes, 7.04%), the oxidative phosphorylation (ko00190, 413 unigenes, 3.15%), the purine metabolism (ko00230, 372 unigenes, 2.84%), and the protein processing in the endoplasmic reticulum (ko04141, 317 unigenes, 2.42%).
Figure 3. Classification of clusters of orthologous groups (COG).
Candidate enzymes involved in the biosynthesis of cellulose and lignin
Cellulose and lignin, which account for most of the dry weight of wood, are two important biopolymers. We focused the next step of our analysis on the genes involved in the biosynthesis of cellulose and lignin.
At present, cellulose synthesis is one of the main fields in plant molecular biology research. Based on previous studies [Citation29,Citation30], we identified 111 unigenes associated with 6 enzymes of the cellulose biosynthesis pathway in the annotated P. neoveitchii transcriptome (). The cellulose synthase complex (CSC) is composed of cellulose synthase catalytic subunits (CesA). In this study, 59 unigene sequences were annotated as encoding CesA subunits.
Figure 4. Number of P. neoveitchii unigenes that may be involved in the biosynthesis pathways of cellulose (A) and lignin (B).
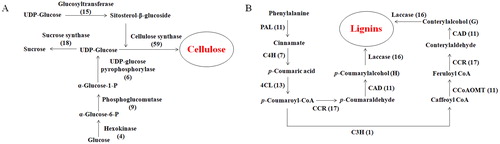
Although lignin research has been carried out for many years, some aspects of the lignin biosynthesis remain a debated topic. In this study, we identified 105 assembled unigenes which were associated with 10 enzymes in the normal phenylpropane pathway ().
However, since cellulose and lignin biosynthesis are complex processes involving multiple parameters, we assume that our transcriptome sequencing analysis will throw light on only a part of the whole process. To reach more specific conclusions, we need additional accurate molecular, genomics and proteomics analysis programmes to validate and further build on our predictions.
Identification and analysis of transcription factors in P. neoveitchii
Many important traits of woody plants are of particular interest to researchers, such as growth, development and stress responses. These traits are tightly dependent on transcription factors (TFs) such as MYB, WRKY, bHLH, WD40, NAC, MADS-box, GATA, CCAAT, bZIP and AP2/ERF.
Based on the sequence and annotation information from NR, NT, SWISS-PROT, GO and the KEGG database, 514 proteins were identified as putative TFs that belong to different TF families (). In the model plant species Arabidopsis thaliana, the MYB family is one of the largest TF families [Citation31]. In our dataset, the most abundant family was also the MYB family. Being the largest TF groups in plants, the MYB family has demonstrated its essential role for the responses to abiotic stresses [Citation32–35]. In addition, the WRKY, bHLH and NAC TFs were also identified in the dataset, and efforts have been made to unveil the molecular mechanism of the responses to abiotic stresses in Arabidopsis, Oryza sativa, Populus and other plant species [Citation36–41]. Increasing evidence has been provided for the crucial functions of TFs, as they relate to mediating the growth and development of plants. This evidence increases our knowledge of the functional importance of the MADS-box genes involved in flower, fruit, and seed development [Citation42–48]. Additionally, there have been strides in conifer tree research over the past decade [Citation49–51].
Figure 5. Putative transcription factors encoded by unigenes in P. neoveitchii.
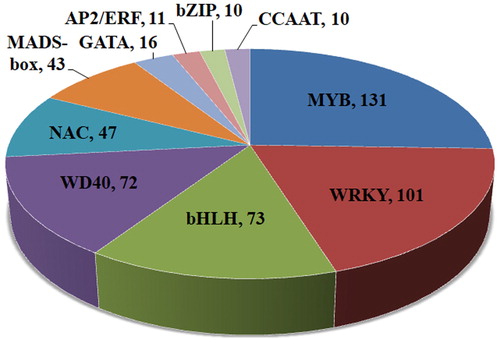
Among the other classes of TFs in Arabidopsis, the transcription factor MYBL2 is a transcriptional repressor that regulates anthocyanin biosynthesis by modulating the MYB/bHLH/WD40 complex [Citation52]. Similarly, AtMYB15 combines with the bHLH protein AtICE1, and they both bind to the AtCBF3 promoter to regulate the cold stress response [Citation53]. Overall, through in-depth analysis of transcription factors in our transcriptome dataset, we can identify candidate genes involved in the growth and development, plant signalling pathways and regulatory networks for this conifer tree.
EST-SSR markers discovery
The SSR markers are the most widely used molecular marker systems. To explore the EST-SSR markers, we extracted 1,560 sequences containing 1,814 EST-SSRs from 12,845 unigenes. The most highly represented repeat units of potential SSRs were five and six, which accounted for 46.67% (414) and 29.43% (261), respectively (). The most common repeat sequences were AT/AT (156), AGC/GCT (132), AAG/CTT (119) and AGG/CCT (112) (Supplemental Table S3). We believe that this large set of EST-SSR markers in this study may provide new resource for genetics, evolution and breeding research in this tree species.
Table 3. Length distribution of EST-SSRs based on the number of repeat units.
Conclusions
To the best of our knowledge, this study provides the first comprehensive transcriptome of P. neoveitchii. The coverage of the transcriptome includes 12.97 Gbp, with a total of 66,022 different unigenes. We used the assembled transcriptome to identify enzymes involved in the biosynthesis of cellulose and lignin and transcription factors involved in stress responses, growth and development. We also proposed a large number of EST-SSR markers, which will be helpful for the subsequent population genetic studies and molecular breeding.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Cai J, Yang XP, Jiang ZM. Researches on the state key conservation plant species in zhouzhi national-level nature reserve. J Northwest Forest Univ. 2014;17:63–66.
- Zhang DS, Yongshik K, Maunder M, et al. The conservation status and conservation strategy of Picea neoveitchii. Chin J Popul Resource Environ. 2006;4:58–64.
- Chen WQ, Song ZJ, Xu HH. A new antifungal and cytotoxic C-methylated flavone glycoside from Picea neoveitchii. Bioorg Med Chem Lett. 2012;22:5819–5822.
- Song Z, Chen W, Du X, et al. Chemical constituents of Picea neoveitchii. Phytochemistry. 2011;72:490–494.
- Li L, Zhang H, Liu Z, et al. Comparative transcriptome sequencing and de novo analysis of Vaccinium corymbosum during fruit and color development. BMC Plant Biol. 2016;16:223–232.
- Ma T, Wang J, Zhou G, et al. Genomic insights into salt adaptation in a desert poplar. Nat Commun. 2013;4:2797–2806.
- Qi X, Li MW, Xie M, et al. Identification of a novel salt tolerance gene in wild soybean by whole-genome sequencing. Nat Commun. 2014;5:1–11.
- Xiao L, Yang G, Zhang L, et al. The resurrection genome of Boea hygrometrica: A blueprint for survival of dehydration. Proc Natl Acad Sci USA. 2015;112:5833–5837.
- Dai X, Hu Q, Cai Q, et al. The willow genome and divergent evolution from poplar after the common genome duplication. Cell Res. 2014;24:1274–1277.
- He N, Zhang C, Qi X, et al. Draft genome sequence of the mulberry tree Morus notabilis. Nat Commun. 2013;4:1–9.
- Liu MJ, Zhao J, Cai QL, et al. The complex jujube genome provides insights into fruit tree biology. Nat Commun. 2014;5:1–12.
- Myburg AA, Grattapaglia D, Tuskan GA, et al. The genome of Eucalyptus grandis. Nature. 2014;510:356–362.
- Tuskan GA, Difazio S, Jansson S, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science. 2006;313:1596–1604.
- Velasco R, Zharkikh A, Affourtit J, et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat Genet. 2010;42:833–839.
- Verde I, Abbott AG, Scalabrin S, et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet. 2013;45:487–494.
- Birol I, Raymond A, Jackman SD, et al. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics. 2013;29:1492–1497.
- Nystedt B, Street NR, Wetterbom A, et al. The Norway spruce genome sequence and conifer genome evolution. Nature. 2013;497:579–584.
- Zimin A, Stevens KA, Crepeau MW, et al. Sequencing and assembly of the 22-gb loblolly pine genome. Genetics. 2014;196:875–890.
- Kuzmin DA, Feranchuk SI, Sharov VV, et al. Stepwise large genome assembly approach: a case of Siberian larch (Larix sibirica Ledeb. ). BMC Bioinformatics. 2019;20:36–52.
- Zimin AV, Stevens KA, Crepeau MW, et al. An improved assembly of the loblolly pine mega-genome using long-read single-molecule sequencing. Gigascience. 2017;6:1–4.
- Guan R, Zhao Y, Zhang H, et al. Draft genome of the living fossil Ginkgo biloba. Gigascience. 2016;5:49–63.
- Englehardt J. Sources, industrial derivatives, and commercial applications of cellulose. Carbohydr Eur. 1995;12:5–14.
- Kieber JJ, Polko J. The regulation of cellulose biosynthesis in plants. Plant Cell. 2019;31:282–296.
- Moura JC, Bonine CA, de Oliveira Fernandes Viana J, et al. Abiotic and biotic stresses and changes in the lignin content and composition in plants. J Integr Plant Biol. 2010;52:360–376.
- Ohtani M, Demura T. The quest for transcriptional hubs of lignin biosynthesis: beyond the NAC-MYB-gene regulatory network model. Curr Opin Biotechnol. 2019;56:82–87.
- Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652.
- Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Meth. 2008;5:621–628.
- Conesa A, Götz S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genom. 2008;2008:1–12.
- Djerbi S, Lindskog M, Arvestad L, et al. The genome sequence of black cottonwood (Populus trichocarpa) reveals 18 conserved cellulose synthase (CesA) genes. Planta. 2005;221:739–746.
- Guerriero G, Fugelstad J, Bulone V. What do we really know about cellulose biosynthesis in higher plants? J Integr Plant Biol. 2010;52:161–175.
- Feng G, Burleigh JG, Braun EL, et al. MYB gene family in plants. Genome Biol Evol. 2017;9:1013–1029.
- Cui MH, Yoo KS, Hyoung S, et al. An Arabidopsis R2R3-MYB transcription factor, AtMYB20, negatively regulates type 2C serine/threonine protein phosphatases to enhance salt tolerance. FEBS Lett. 2013;587:1773–1778.
- Dai X, Wang Y, Yang A, et al. OsMYB2P-1, an R2R3 MYB transcription factor, is involved in the regulation of phosphate-starvation responses and root architecture in rice. Plant Physiol. 2012;159:169–183.
- Seo PJ, Lee SB, Suh MC, et al. The MYB96 transcription factor regulates cuticular wax biosynthesis under drought conditions in Arabidopsis. Plant Cell. 2011;23:1138–1152.
- Shin D, Moon SJ, Han S, et al. Expression of StMYB1R-1, a novel potato single MYB-like domain transcription factor, increases drought tolerance. Plant Physiol. 2011;155:421–432.
- De Clercq I, Vermeirssen V, Van Aken O, et al. The membrane-bound NAC transcription factor ANAC013 functions in mitochondrial retrograde regulation of the oxidative stress response in Arabidopsis. Plant Cell. 2013;25:3472–3290.
- Ishihama N, Yamada R, Yoshioka M, et al. Phosphorylation of the Nicotiana benthamiana WRKY8 transcription factor by MAPK functions in the defense response. Plant Cell. 2011;23:1153–1170.
- Jiang Y, Duan Y, Yin J, et al. Genome-wide identification and characterization of the Populus WRKY transcription factor family and analysis of their expression in response to biotic and abiotic stresses. J Exp Botany. 2014;65:6629–6644.
- Jiang Y, Liang G, Yu D. Activated expression of WRKY57 confers drought tolerance in Arabidopsis. Mol Plant. 2012;5:1375–1388.
- Lee S, Seo PJ, Lee HJ, et al. A NAC transcription factor NTL4 promotes reactive oxygen species production during drought-induced leaf senescence in Arabidopsis. Plant J. 2012;70:831–844.
- Xu ZY, Kim SY, Hyeon do Y, et al. The Arabidopsis NAC transcription factor ANAC096 cooperates with bZIP-type transcription factors in dehydration and osmotic stress responses. Plant Cell. 2013;25:4708–4724.
- Chen MK, Hsu WH, Lee PF, et al. The MADS box gene, FOREVER YOUNG FLOWER, acts as a repressor controlling floral organ senescence and abscission in Arabidopsis. Plant J. 2011;68:168–185.
- Deng W, Ying H, Helliwell CA, et al. FLOWERING LOCUS C (FLC) regulates development pathways throughout the life cycle of Arabidopsis. Proc Natl Acad Sci USA. 2011;108:6680–6685.
- Garay-Arroyo A, Ortiz-Moreno E, de la Paz Sanchez M, et al. The MADS transcription factor XAL2/AGL14 modulates auxin transport during Arabidopsis root development by regulating PIN expression. EMBO J. 2013;32:2884–2895.
- Sang X, Li Y, Luo Z, et al. CHIMERIC FLORAL ORGANS1, encoding a monocot-specific MADS box protein, regulates floral organ identity in rice. Plant Physiol. 2012;160:788–807.
- Smaczniak C, Immink RG, Angenent GC, et al. Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development. 2012;139:3081–3098.
- Albert VA, Barbazuk WB, dePamphilis CW, et al. The Amborella genome and the evolution of flowering plants. Science. 2013;342:1467–1477.
- Uddenberg D, Reimegard J, Clapham D, et al. Early cone setting in Picea abies acrocona is associated with increased transcriptional activity of a MADS box transcription factor. Plant Physiol. 2013;161:813–823.
- Wegrzyn JL, Lee JM, Tearse BR, et al. TreeGenes: A forest tree genome database. Int J Plant Genom. 2008;2008:1–7.
- Pireyre M, Burow M. Regulation of MYB and bHLH transcription factors: a glance at the protein level. Mol Plant. 2015;8:378–388.
- Nemesiogorriz M, Blair PB, Dalman K, et al. Identification of Norway spruce MYB-bHLH-WDR transcription factor complex members linked to regulation of the flavonoid pathway. Front Plant Sci. 2017; [cited 2019 May 02]8:305.
- Zhang H, Forde BG. An Arabidopsis MADS box gene that controls nutrient-induced changes in root architecture. Science. 1998;279:407–409.
- Agarwal M, Hao Y, Kapoor A, et al. A R2R3 type MYB transcription factor is involved in the cold regulation of CBF genes and in acquired freezing tolerance. J Biol Chem. 2006;281:37636–37645.