
Abstract
4-Hydroxynonenal (HNE), a major end-product of free-radical activated peroxidation of polyunsaturated fatty acids, has attracted great scientific interest. HNE is more stable than free radicals and can diffuse within the cell or leave it and react with targets far from the initial site. These reactive aldehyde species are considerably reactive, producing multiple intra- and inter-molecular covalent adducts with biomolecules such as proteins, DNA and phospholipids. HNE is the most intensively studied aldehyde in relation to its physiological and protective function as a signalling molecule that stimulates gene expression and cell survival, as well as for its pathophysiological role as a toxic messenger that can propagate and amplify oxidative injury and promote mitochondrial dysfunction and cell death. Non-alcoholic fatty liver disease (NAFLD) is the most prevalent form of chronic liver disease in the world associated with oxidative stress, mitochondrial dysfunction and hepatocellular apoptosis. In this review we focus our attention on the molecular mechanism of the signalling and regulatory action of HNE. The role of HNE as a potent mediator for progression of liver injury in NAFLD is also discussed.
Introduction
Among the 4-hydroxyalkenals, 4-hydroxy-2-nonenal (HNE) is the most studied compound. Both, physiological and toxic roles of HNE have been reported [Citation1–3]. HNE derives from oxidation of membrane n-6-polyunsaturated fatty acids (PUFAs), essentially arachidonic acid and linoleic acid, two of the most important fatty acids in membranes. HNE is amphiphilic; it is water soluble but displays strong lipophilic characteristics. Consequently, the aldehydes tend to settle in biomembranes where phospholipids, such as phosphotydil-ethanolamine, and proteins like transporters, ion channels and receptors, rapidly interact with HNE [Citation4]. At low concentrations in human tissues and plasma, HNE is involved in the control of signal transduction, cell proliferation, differentiation and cell cycle regulation [Citation5–9]. In addition, these compounds may also play the role of signalling molecules that directly amend cell functions in a hormetic manner in order to enable cells to adapt to stressful stimuli [Citation10].
At high concentrations, HNE exhibits toxic effects of lipid peroxidation. These aldehydes contribute to depletion of cellular sulphhydril compounds such as glutathione (GSH), which changes the redox status. Consequently, HNE has been commonly acknowledged as an inducer and mediator of oxidative stress [Citation11]. The toxicity of aldehydes results from alterations in a number of cell functions which predominantly depend on the formation of covalent adducts with cellular proteins [Citation8]. The amphiphilic features of aldehydes contribute to their diffusion across membranes, which later leads to covalent modification of the proteins in the cytoplasm and nucleus [Citation12]. In addition to the plasma membranes, both mitochondria and the endoplasmic reticulum are markedly susceptible to HNE-induced damage [Citation7,Citation13–15]. HNE is not only a marker of oxidative stress but can act as a mediator of oxidative damage by modifying specific cellular functions [Citation16]. Therefore, HNE is considered to be a toxic messenger that can deepen and expand oxidative damage in cells [Citation8].
HNE reactivity and metabolism
HNE has high reactivity and interacts easily with thiol (-SH) or amino (-NH2) groups of glutathione and proteins, and at high concentration, with DNA. HNE forms covalent adducts with proteins in the cell, including ones in the plasma membrane, with the modification of specific cellular functions [Citation16,Citation17]. Plasma membrane proteins represent the first targets for adduct formation. HNE can also interact with nuclear proteins, hence modifying protein expression via their interaction with transcription factors or other regulatory compounds [Citation7,Citation8]. Functional and or signalling proteins modulation probably represents one of the main mechanism by which HNE can modify both physiological and pathological processes [Citation18] ().
Figure 1. Reactivity, metabolism and possible biological effects of 4-hydroxynonenal.
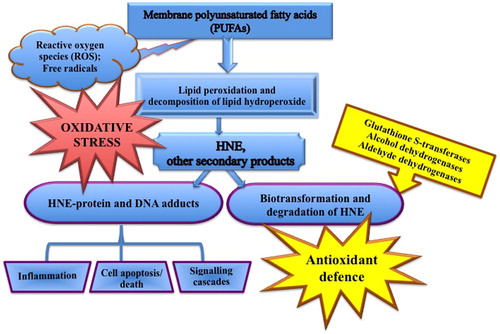
As recently reviewed [Citation17], HNE-protein adducts are physiological products of metabolism which are sometimes elevated in tissues or plasma in diseases [Citation1]. HNE protein adducts at low concentrations play an important role in cell signal transduction and gene expression including receptors, kinases, phosphatases and transcription factors [Citation7,Citation19,Citation20].
The high reactivity that HNE has leads to an array of effects on various proteins, resulting in changes in their function and stability. High levels of HNE may influence cellular function and behaviour [Citation17]. The formation of cross-linked HNE-protein adducts might notably affect the cellular senescence process and, thus, contribute to aging [Citation3,Citation17,Citation20]. Studies have shown that the generation of HNE-protein adducts can play important pathogenic roles in age-related neurodegenerative diseases, atherosclerosis and cancer [Citation21–24]. Many animal model studies and clinical trials have affirmed that HNE modified proteins are one of the important factors in the development and progression of chronic liver diseases [Citation25,Citation26].
The generation of HNE-protein adducts can represent a contrast to the progression of disease or can promote adaptive cell responses, demonstrating that HNE is not only a toxic product of LPO but also a regulatory molecule that is involved in several biochemical pathways [Citation4,Citation26]. While high concentrations of lipid peroxidation products such as 4-HNE cause cellular damage and apoptosis, in the low concentration range, HNE exerts adaptive cytoprotective effect. In other words, the effect of 4-HNE on the cell may be damaging or protective, depending on its concentration, respectively, not only on its production but also on the clearance of this molecule [Citation2]. This suggests that, in some diseases, HNE may act as a target for detection, early prevention, and even cessation of disease progression [Citation27].
The degradation of HNE is an important part of the antioxidant defense. HNE is degraded to non-toxic or less toxic compounds which are either metabolised or excreted [Citation17]. The key HNE-degrading enzymes are glutathione S-transferases (GST), alcohol dehydrogenases (ADH) and aldehyde dehydrogenases (ALDH) [Citation3,Citation17,Citation28]. In rat hepatocytes, more than half of HNE is metabolised to the GSH-HNE intermediate conjugate [Citation17,Citation29]. GSH-HNE and other intermediates are among the main determinants of the intracellular levels of HNE because they have the ability to modulate stress-induced signalling for programmed cell death. These processes, in turn, limit the apoptotic potential of HNE [Citation2,Citation20,Citation30]. HNE detoxification by GSTs can be reduced due to depletion of GSH, which in turn may lead to increased levels of HNE [Citation31,Citation32]. On the other hand, S-adenosyl methionine (SAM) diminishes HNE modification of enzymes which have a vital role in the detoxification of HNE [Citation32]. N-acetyl cysteine (NAC) is a precursor of GSH synthesis and a physiological HNE scavenger, which may directly neutralise HNE via its antioxidant and aldehyde scavenger properties [Citation33].
HNE in signalling pathways
The most widely described roles of HNE in the signalling pathways are associated with its activation of kinases, as well as transcription factors that are responsible for redox homeostasis (Ref-1, Nrf2, p53, NFκB) [Citation34]. Depending on its level, HNE exerts harmful or protective effects associated with the induction of antioxidant defense mechanisms [Citation34].
Oxidative stress and lipid peroxide HNE may activate Nuclear Factor-κB (NF-κB) a redox-sensitive transcription factor that regulates expression of numerous genes and plays important roles in immune and stress responses, inflammation, and apoptosis [Citation35,Citation36]. NF-kB is sequestered in the cytoplasm of cells and is bound it inhibitor IkB. NF-kB is activated by degradation of inhibitor by HNE and inflammatory cytokines [Citation3]. After liberation, NF-kB translocate to the nucleus binding to DNA and enhances the expression of genes TNF-α, IL-1β, Il-6 and IL-8 and consequently lead to mtDNA damage which results in generation of more reactive oxygen species (ROS), forming a vicious cycle of oxidative stress [Citation37]. Therefore, antioxidants that inhibit the activation of NF-kB may be attractive strategy for limiting hepatic injury in NAFLD [Citation38,Citation39]. NF-kB and activator protein 1 (AP1) are downstream signalling molecules that couple receptor ligation to the activities of several classes of signal-dependent transcription factors, such as c-Jun NH2-terminal kinase (JNK) [Citation40,Citation41].
HNE could act as a potential activator of Nuclear factor erythroid 2–related factor 2 (Nrf2) [Citation3,Citation42,Citation43]. The mechanism involves stress-generated electrophiles and oxidants that switch on the Nrf2 dependent cellular defense pathway. In this defense pathway, Nrf2 is released from Keap1 and translocated to the nucleus where it binds to the antioxidant response element (ARE) and initiates the transcription of antioxidant and protective genes [Citation34,Citation44,Citation45]. Many antioxidant enzymes are regulated by Nrf2, including: NAD(P)H quinone reductase 1, which neutralises the toxicity of quinones; glutamate-cysteine ligase, which regulates GSH synthesis; sulfiredoxin one and thioredoxin reductase 1, which help to detoxify peroxides, hydrogen peroxide and peroxynitrite; heme oxygenase-1 (HO-1), which catalyses the degradation of heme; GST, the cystine/glutamate amino acid transporter and other protective systems [Citation44,Citation45]. Therefore, HNE activating Nrf2 can induce antioxidant defense mechanisms to restrain its own production and to enhance the cellular protection against oxidative stress [Citation22,Citation43]. The role of Nrf2 in the progression of NASH has been studied [Citation46]. It was found that high fat diet induced an increase in lipid peroxidation and Nrf2 expression, which strongly correlated with the degree of hepatic steatosis and inflammation [Citation47]. In mice with diet-stimulated nonalcoholic steatohepatitis, pharmacologic activation of transcription factor Nrf2 improves glucose homeostasis and inhibits hepatic steatosis, inflammation, and fibrosis. Nrf2-mediated amelioration of nonalcoholic steatohepatitis and liver fibrosis involves downregulation of lipogenic genes, induction of antioxidant genes, and suppression of both oxidative and endoplasmic reticulum stress [Citation48].
HNE and apoptosis
The role of HNE in apoptosis has been reviewed more extensively previously [Citation20]. Briefly, what makes HNE a special apoptotic inducer is that it forms protein adducts in mitochondria, causing mitochondrial dysfunction, and propagates oxidative injury [Citation1,Citation14,Citation20,Citation23]. HNE can directly induce apoptosis but it can also be a mediator of apoptosis, as it can be generated by ROS [Citation14,Citation20]. HNE can stimulate intrinsic and extrinsic apoptotic pathways and interact with typical actors such as JNK, Fas or mitochondrial regulators [Citation6,Citation12,Citation15,Citation20,Citation22,Citation49].
HNE induces the intrinsic apoptotic pathway. The intrinsic apoptotic pathway, also called the mitochondrial pathway, is the major apoptotic pathway [Citation20]. ROS and lipid-peroxides products such as 4-HNE directly damage mitochondrial (mt) DNA and activate the mitochondrial apoptotic pathway. Oxidation of cardiolipin (the major lipid component of the inner mitochondrial membrane), which generates HNE, is necessary for the subsequent steps of intrinsic apoptosis [Citation50,Citation51]. In addition, HNE oxidises the SH-groups (cysteine, methionine) of glutathione, which changes the redox status and is an early and potent activator of apoptotic signals [Citation13,Citation14,Citation49,Citation52,Citation53]. According to Liu et al. [Citation52], HNE-induced reduction of cellular GSH/GSSG pool is closely associated with the activity of HNE to induce apoptotic cell death via Fas-independent activation of caspase 3, possibly through a mitochondria-dependent pathway.
Mitochondria are very sensitive to the damaging effect of HNE. Damage to mtDNA by HNE causes defects in respiratory enzymes, decreases the capacity of reducing potentials and energy production. On the one hand, it further enhances the generation of ROS and reactive nitrogen species (RNS) in mitochondria, resulting in disturbance in antioxidant defense [Citation8,Citation53]. This disruption acts as an apoptotic signal [Citation20]. Under the effect of an apoptotic signal (mtDNA damage), the pro-apoptotic Bax proteins undergoing conformational changes, are translocated from the cytosol to the external mitochondrial membrane where the anti-apoptotic Bcl-2 proteins are located. The pro-apoptotic protein Bax increases the permeability of the external mitochondrial membrane, which allows the release of cytochrome C and other pro-apoptotic molecules in the cytosol and activation of caspases and the mitochondrial pathway of apoptosis [Citation54–56].
HNE has a significantly different role compared to other lipid oxidation products in the regulation of two essential anti-apoptotic elements, the NF-κB transcription factor and its target anti-apoptotic B-Cell Lymphoma-2 (Bcl-2) protein. There is evidence for presence of a HNE-based downregulation of Bcl-2 expression, mostly likely via NF-κB. The crosstalk of NF-κB pathway and Bcl-2 was proposed to be mediated by HNE [Citation36].
HNE induces the extrinsic apoptotic pathway. The extrinsic apoptotic pathway transmits a death signal from the cell surface to the intracellular compartments and is initiated by the activation of death receptors like receptors of TNF-α, Fas/CD95 or TRAIL (TNF-related apoptosis-inducing ligand). The induction of Fas by HNE is associated with JNK activation and apoptosis induction [Citation57]. JNK is known to have a central role in both intrinsic and extrinsic pathways. Activation of JNK pathways and production of reactive oxygen species (ROS) from fructose metabolism can stimulate the production of inflammatory mediators (via NF-kB) or induce direct apoptosis. On the other hand, apoptosis may also be activated by the external receptor-dependent pathway via Fas ligand, TNFR1 and TRALL receptors [Citation58]. It has been shown that elevated serum TNF-α levels [Citation3] and saturated CMKs can induce apoptosis by binding to the corresponding membrane receptors in a ‘death-inducing signal complex’ activating the catalytic activity of effector caspases [Citation59,Citation60]. HNE also triggers NF-κB activation, which then induces TNF-α synthesis and consequently mtDNA damage.
Stimulation of the death receptors by TNFα may also increase the expression of the pro-apoptotic Bax in the external mitochondrial membrane and the release of cytochrome C, thereby binding the death receptor and mitochondrial pathway [Citation61,Citation62].
The pro-apoptotic activity of HNE as a marker of lipid peroxidation and oxidative stress is associated with: (i) direct damage and mtDNA activation of the mitochondrial apoptotic pathway; (ii) inhibition of mitochondrial proteins (channels) such as adenine nucleotide translocase (ANT) due to SH-group oxidation, which increases the permeability of mitochondrial membrane for Cyt-c release; (iii) inhibition of antiapoptotic Bcl-2 [Citation63].
HNE and liver injury in NAFLD
Diets rich in fat, as well as high-fructose diets, and high-cholesterol diets contribute to the pathogenesis of nonalcoholic liver disease (NAFLD) [Citation64]. This disease constitutes an increasingly prevalent liver disorder and has been suggested to be the hepatic manifestation of the metabolic syndrome [Citation17,Citation65]. NAFLD is a clinical-pathological syndrome that encompasses a wide spectrum of liver damages, ranging from steatosis to non-alcoholic steatohepatitis (NASH) and cirrhosis in the absence of alcohol abuse [Citation66]. The pathogenesis of this disease is multifactorial and not fully elucidated. Studies have shown that fat accumulation in liver cells, imbalance in adipocytokines secretion, oxidative stress and low-grade inflammation, are responsible for the development of mitochondrial dysfunction, stimulating hepatocyte apoptosis in the steatotic liver [Citation67,Citation68].
The mechanisms by which steatosis progresses to NASH are still not fully understood; however, growing evidence has suggested that oxidative stress is involved in this process [Citation69–73]. Oxidative stress is generated through oxidation of free fatty acids, cytochrome P450, iron overload, and necro-inflammatory cytokines such as TNF-α [Citation74–76]. Ensuing excessive ROS production enhances lipid peroxidation, which subsequently decreased the antioxidant defense, activates lipid peroxidation and elevates the intracellular and extracellular content of 4-HNE, and results in hepatocyte damage and liver injury [Citation17,Citation25]. However, the cellular and molecular mechanisms of HNE-induced liver injury associated with chronic fructose and lipid consumption have not been well elucidated yet. Some of the most relevant recent findings of HNE as bioactive marker of oxidative stress are summarised in .
Table 1. Reports of HNE as a bioactive marker of oxidative stress.
Chronic fructose consumption causes fat accumulation in the liver and liver steatosis [Citation77]. Surplus of fatty acids in fatty liver leads to mitochondrial production of free radicals which may stimulate inflammatory response and liver injury in NAFLD. Extra-mitochondrial reactions mediated by NADPH oxidase, xanthine oxidase and inducible nitric oxide synthase (iNOS) may contribute to the elevated ROS/RNS production in NAFLD [Citation78,Citation79]. Elevated oxidative stress has been well documented in NAFLD patients [Citation17,Citation73,Citation80]. Excessive ROS production overwhelms antioxidant defenses and generates highly toxic HNE lipid peroxides. Significant increase in serum levels of HNE levels has been demonstrated in patients with NASH, compared to those with simple steatosis [Citation81]. An excessive generation of HNE detected by antibodies, like anti-HNE protein, was demonstrated in liver in experimental models of NAFLD. In NAFLD, however, parallel with the increase of mitochondrial ROS production, diminished expression and activity of ROS detoxification mechanisms (e.g. SOD2, catalase or GSH were found [Citation79,Citation82,Citation83]. Lower hepatic expression of GST, the main detoxifying enzyme of HNE, was reported in obese patients with steatosis compared to obese individuals without NAFLD [Citation84]. Thus, low levels of GSH and decreased expression and/or activity of some glutathione-related enzymes may have additional deleterious effects on fatty liver [Citation52,Citation80,Citation85].
Mitochondrial dysfunction and oxidative stress play a central role in the pathophysiology of NAFLD/NASH [Citation86]. Mitochondria are very sensitive to the damaging action of lipid peroxide HNF, which oxidise SH-groups (cysteine, methionine) of glutathione, change the redox status and are an early and powerful activator of apoptotic signals [Citation70]. The formation of HNE adducts, GSH depletion and protein oxidation are major events associated with mitochondrial dysfunction, which represent critical initiating events for the development of NAFLD [Citation83]. HNE is a special apoptotic inducer because of its abilities to oxidise mitochondrial DNA, protein and to form protein adducts [Citation20]. A significant increase in mitochondrial HNE–protein adducts during NASH development has been also reported [Citation68–72]. In addition, oxidation of biomolecules by mitochondrial ROS initiates a vicious cycle of exacerbated mitochondrial dysfunction and cell death [Citation79].
HNE level is elevated in steatosis liver, indicating oxidative damage of mitochondria. In rats with fructose-induced metabolic syndrome the expression of pro-apoptotic BAX in the liver is increased, whereas that of Bcl-2 decreased. Similar findings for apoptotic proteins Вax and Bcl-2 in SECs and hepatocytes are demonstrated in an experimental model of NAFLD [Citation87]. Under physiological conditions Bcl-2 located in the mitochondrial membrane suppresses oxidative stress and prevents cell apoptosis [Citation88]. The activation of oxidative stress by TNF-α and free fatty acids translocation of the BAX protein from the cytosol and its transfer to the external mitochondrial membrane result in leakage of cytochrome C and other pro-apoptotic proteins and activation of programmed cell death [Citation14]. Depletion of mitochondrial GSH levels could also be due to its reduced uptake by mitochondria as a result of enhanced levels of cholesterol within the inner mitochondrial membrane and the decrease in synthesis of SAM, the major methyl donor in liver and precursor to GSH [Citation89]. An increased HNE adduct level and GSH deficiency may promote the opening of mitochondrial permeability transition pores, which is a critical factor for cell death [Citation90]. In mice with diet-stimulated nonalcoholic steatohepatitis, a pharmacologic activator of Nrf2 (Curcumin) prevents the NASH by mitochondria protection and apoptosis reduction and providing a possible novel treatment for NASH [Citation25].
Additionally, lipoperoxide products such HNE increase the production of pro-inflammatory cytokines such as TNF-α, IL-6 and CRP, and contribute to apoptosis and progression of liver injury in NAFLD [Citation91]. Bcl-2 and BAX protein, major mediators of apoptosis, are mainly activated in the internal (mitochondrial) signalling pathway. It has been reported that the stimulation of the death receptors by TNFα may also increase the expression of the pro-apoptotic Bax in the external mitochondrial membrane and may result in release of cytochrome C, thereby binding the death receptor and mitochondrial pathway [Citation61,Citation62,Citation92]. Lipid peroxide HNE in steatotic liver may synergistically activate the JNK/c-Jun signalling pathway that augmentes liver injury and apoptosis [Citation93]. HNE-induced JNK activation is inhibited by pretreatment of the cells with a thiol antioxidant, N-acetylcysteine [Citation41,Citation72,Citation94]. Tsuruta et al. [Citation95] showed JNK-induced Bax translocation to mitochondria, cytochrome c release and apoptosis. Sustained activation of the c-Jun NH2-terminal kinase (JNK) signalling pathway mediates the development and progression of experimental diet-induced NAFLD [Citation95].
Data from a study by Malhi et al. [Citation96] show that saturated fatty acids also induce JNK-dependent hepatocyte apoptosis by activating pro-apoptotic Bid-Bach proteins from the internal signalling pathway. Antioxidant gingolide A increased the levels of anti-apoptotic Bcl-2, while it decreased Bax, phosphorylated JNK and cleaved caspase-3 and -9 levels in the livers of animals with high fat diet [Citation97]. The manifestation of a synergistic effect on the activation of the JNK signalling pathway from depletion of mitochondrial reduced glutathione and elevated peroxides, such as MDA, was found in the steatal liver in experimental animals and patients [Citation74].
HNE is one of the major end-products of lipid peroxidation generated during the transition from simple steatosis to steatohepatitis [Citation72]. Therefore HNE generated by NAFLD is not only a passive biomarker but rather an active mediator of hepatocellular injury [Citation31,Citation93]. Thus HNE may mediate the progression from steatosis to hepatocyte injury by direct oxidative modification of cellular molecules and also by activation of cell death signalling pathways such as JNK/c-Jun.
Conclusions
The increased evidence in liver tissue and plasma definitely supports a significant role of oxidative stress and free-radical-induced HNE production in liver pathophysiology. This aldehyde is not only a toxic byproduct of oxidative stress metabolism but, on the contrary, it is an important factor in the mechanism of cell signalling. Studies with NAFLD rats show pathological HNE features such as steatosis, dysregulated oxidative unbalance, low-grade inflammation, early mitochondrial dysfunction and apoptosis, which can be prevented in the presence of antioxidants.
Disclosure statement
The authors declare no conflict of interest.
References
- Dianzani MU. 4-Hydroxynonenal from pathology to physiology. Mol Aspects Med. 2003;24(4–5):263–272.
- Chen ZH, Niki E. 4-hydroxynonenal (4-HNE) has been widely accepted as an inducer of oxidative stress. Is this the whole truth about it or can 4-HNE also exert protective effects? IUBMB Life. 2006;58(5–6):372–373.
- Zhang H, Forman HJ. Signaling by 4-hydroxy-2-nonenal: exposure protocols, target selectivity and degradation. Arch Biochem Biophys. 2017;617:145–154.
- Poli G, Biasi F, Leonarduzzi G. 4-Hydroxynonenal-protein adducts: a reliable biomarker of lipid oxidation in liver diseases. Mol Aspects Med. 2008;29(1–2):67–71.
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11(1):81–128.
- Awasthi YC, Sharma R, Sharma A, et al. Self-regulatory role of 4-hydroxynonenal in signaling for stress-induced programmed cell death. Free Radic Biol Med. 2008;45(2):111–118.
- Jacobs AT, Marnett LJ. Systems analysis of protein modification and cellular responses induced by electrophile stress. Acc Chem Res. 2010;43(5):673–683.
- Pizzimenti S, Ciamporcero E, Daga M, et al. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front Physiol. 2013;4:242
- Milkovic L, Cipak Gasparovic A, Zarkovic N. Overview on major lipid peroxidation bioactive factor 4-hydroxynonenal as pluripotent growth-regulating factor. Free Radic Res. 2015;49(7):850–860.
- Forman HJ, Fukuto JM, Miller T, et al. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch Biochem Biophys. 2008;477(2):183–195.
- Uchida K. 4-hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42(4):318–334.
- Negre-Salvayre A, Vieira O, Escargueil-Blanc I, et al. Oxidized LDL and 4-hydroxynonenal modulate tyrosine kinase receptor activity. Mol Aspects Med. 2003;24(4–5):251–261.
- Raza H, John A. 4-hydroxynonenal induces mitochondrial oxidative stress, apoptosis and expression of glutathione S-transferase A4-4 and cytochrome P450 2E1 in PC12 cells. Toxicol Appl Pharmacol. 2006;216(2):309–318.
- Roede JR, Jones DP. Reactive species and mitochondrial dysfunction: mechanistic significance of 4-hydroxynonenal. Environ Mol Mutagen. 2010;51(5):380–390.
- Chapple SJ, Cheng X, Mann GE. Effects of 4-hydroxynonenal on vascular endothelial and smooth muscle cell redox signaling and function in health and disease. Redox Biol. 2013;1(1):319–331.
- Arashiki N, Otsuka Y, Ito D, et al. The covalent modification of spectrin in red cell membranes by the lipid peroxidation product 4-hydroxy-2-nonenal. Biochem Biophys Res Commun. 2010;391(3):1543–1547.
- Castro JP, Jung T, Grune T, et al. 4-Hydroxynonenal (HNE) modified proteins in metabolic diseases. Free Radic Biol Med. 2017;111:309–315.
- Petersen DR, Doorn JA. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic Biol Med. 2004;37(7):937–945.
- Ullery JC, Marnett LJ. Protein modification by oxidized phospholipids and hydrolytically released lipid electrophiles: investigating cellular responses. Biochim Biophys Acta. 2012;1818(10):2424–2435.
- Dalleau S, Baradat M, Guéraud F, et al. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013;20(12):1615–1630.
- Shoeb M, Ansari NH, Srivastava SK, et al. 4-hydroxynonenal in the pathogenesis and progression of human diseases. CMC. 2013;21(2):230–237.
- Csala M, Kardon T, Legeza B, et al. On the role of 4-hydroxynonenal in health and disease. Biochim Biophys Acta. 2015;1852(5):826–838.
- Di Domenico F, Tramutola A, Butterfield DA. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic Biol Med. 2017;111:253–261.
- Moreau R, Nguyen BT, Doneanu CE, et al. Reversal by aminoguanidine of the age-related increase in glycoxidation and lipoxidation in the cardiovascular system of Fischer 344 rats. Biochem Pharmacol. 2005;69(1):29–40.
- Wang L, Lv Y, Yao H, et al. Curcumin prevents the non-alcoholic fatty hepatitis via mitochondria protection and apoptosis reduction. Int J Clin Exp Pathol. 2015;8(9):11503–11509.
- Barrera G, Pizzimenti S, Ciamporcero ES, et al. Role of 4-hydroxynonenal-protein adducts in human diseases. Antioxid Redox Signal. 2015;22(18):1681–1702.
- Breitzig M, Bhimineni C, Lockey R, et al. 4-Hydroxy-2-nonenal: a critical target in oxidative stress? Am J Physiol Cell Physiol. 2016;311(4):C537–C543.
- Pham RT, Gardner JL, Gallagher EP. Conjugation of 4-hydroxynonenal by largemouth bass (Micropterus salmoides) glutathione S-transferases. Mar Environ Res. 2002;54(3–5):291–295.
- Singhal SS, Singh SP, Singhal P, Horne D, et al. Antioxidant role of glutathione S-transferases: 4-hydroxynonenal, a key molecule in stress-mediated signaling. Toxicol Appl Pharmacol. 2015;289(3):361–370.
- Awasthi YC, Yang Y, Tiwari NK, et al. Regulation of 4hydroxynonenal-mediated signaling by glutathione S-transferases. Free Radic Biol Med. 2004;37(5):607–619.
- Yang Y, Sharma R, Sharma A, et al. Lipid peroxidation and cell cycle signaling: 4-hydroxynonenal, a key molecule in stress mediated signaling. Acta Biochim Pol. 2003;50(2):319–336.
- Brown JM, Kuhlman C, Terneus MV, et al. S-adenosyl-l-methionine protection of acetaminophen mediated oxidative stress and identification of hepatic 4-hydroxynonenal protein adducts by mass spectrometry. Toxicol Appl Pharmacol. 2014;281(2):174–184.
- Sumida Y, Niki E, Naito Y, et al. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic Res. 2013;47(11):869–880.
- Łuczaj W, Gęgotek A, Skrzydlewska E. Antioxidants and HNE in redox homeostasis. Free Radic Biol Med. 2017;111:87–101.
- Yadav UCS, Ramana KV. Regulation of NF-κB-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxid Med Cell Longev. 2013;2013:1.
- Timucin AC, Basaga H. Pro-apoptotic effects of lipid oxidation products: HNE at the crossroads of NF-κB pathway and anti-apoptotic Bcl-2. Free Radic Biol Med. 2017;111:209–218.
- Nagakawa Y, Williams GM, Zheng Q, et al. Oxidative mitochondrial DNA damage and deletion in hepatocytes of rejecting liver allografts in rats: role of TNF-alpha. Hepatology 2005;42(1):208–215.
- Sreeja S, Geetha R, Priyadarshini E, et al. Substitution of soy protein for casein prevents oxidative modification and inflammatory response induced in rats fed high fructose diet. ISRN Inflamm. 2014;2014:1.
- Tian Y, Ma J, Wang W, et al. Resveratrol supplement inhibited the NF-κB inflammation pathway through activating AMPKα-SIRT1 pathway in mice with fatty liver. Mol Cell Biochem. 2016;422(1–2):75–84.
- Huang W, Glass CK. Nuclear receptors and inflammation control: molecular mechanisms and pathophysiological relevance. ATVB. 2010;30(8):1542–1549.
- Uchida K, Shiraishi M, Naito Y, et al. Activation of stress signaling pathways by the end product of lipid peroxidation 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J Biol Chem. 1999;274(4):2234–2242.
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47(1):89–116.
- Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol. 2011;85(4):241–272.
- Zhang H, Davies KJA, Forman HJ. Oxidative stress response and Nrf2 signaling in aging. Free Rad Biol Med. 2015;88(Pt B):314–336.
- Loboda A, Damulewicz M, Pyza E, et al. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. 2016;73(17):3221–3247.
- Bataille AM, Manautou J. Nrf2: a potential target for new therapeutics in liver disease. Clin Pharmacol Ther. 2012;92(3):340–348.
- Cherrington N. Diversity in antioxidant response enzymes in progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos. 2010;38:2293–2301.
- Sharma RS, Harrison DJ, Kisielewski D, et al. Experimental nonalcoholic steatohepatitis and liver fibrosis are ameliorated by pharmacologic activation of Nrf2 (NF-E2 p45-related factor 2). Cell Mol Gastroenterol Hepatol. 2018;5(3):367–398.
- Chaudhary P, Sharma R, Sharma A, et al. Mechanisms of 4-hydroxy-2-nonenal induced pro-and anti-apoptotic signaling. Biochemistry 2010;49(29):6263–6275.
- Liu W, Porter NA, Schneider C, et al. Formation of 4-hydroxynonenal from cardiolipin oxidation: intramolecular peroxyl radical addition and decomposition. Free Rad Biol Med. 2011;50(1):166–178.
- Paradies G, Paradies V, Ruggiero FM, et al. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. WJG. 2014;20(39):14205–14218.
- Liu W, Kato M, Akhand AA, et al. 4-hydroxynonenal induces a cellular redox status-related activation of the caspase cascade for apoptotic cell death. J Cell Sci. 2000;113(Pt 4):635–641.
- Ott M, Gogvadze V, Orrenius S, et al. Mitochondria, oxidative stress and cell death. Apoptosis 2007;12(5):913–922.
- Le Bras M, Clément MV, Pervaiz S, et al. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol Histopathol. 2005;20(1):205–219.
- Valko M, Leibfritz D, Moncol J, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1):44–84.
- Rector RS, Thyfault JP, Uptergrove GM, et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol. 2010;52(5):727–736.
- Sharma R, Sharma A, Dwivedi S, et al. 4-Hydroxynonenal self limits Fas-mediated DISC independent apoptosis by promoting export of Daxx from nucleus to cytosol and its binding to Fas. Biochemistry 2008;47(1):143–156.
- Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003;125(2):437–443.
- Schwabe RF, Uchinami H, Qian T, et al. Differential requirement for c-Jun NH2-terminal kinase in TNFalpha- and Fas-mediated apoptosis in hepatocytes. FASEB J. 2004;18(6):720–722.
- Zelová H, Hošek J. TNF-α signalling and inflammation: interactions between old acquaintances. Inflamm Res. 2013;62(7):641–651.
- Yin XM. Signal transduction mediated by bid, a pro-death Bcl-2 family proteins, connects the death receptor and mitochondria apoptosis pathways. Cell Res. 2000;10(3):161–167.
- Shuh M, Bohorquez H, Loss GE, Jr, et al. Tumor necrosis factor-α: life and death of hepatocytes during liver ischemia/reperfusion injury. Ochsner J. 2013;13(1):119–130.
- Vieira HL, Belzacq AS, Haouzi D, et al. The adenine nucleotide translocator: a target of nitric oxide, peroxynitrite, and 4-hydroxynonenal. Oncogene 2001;20(32):4305–4316.
- Jensen VS, Hvid H, Damgaard J, et al. Dietary fat stimulates development of NAFLD more potently than dietary fructose in sprague-dawley rats. Diabetol Metab Syndr. 2018;10(1):4.
- Katsiki N, Mikhailidis DP, Mantzoros CS. Non-alcoholic fatty liver disease and dyslipidemia: an update. Metabolism 2016;65(8):1109–1123.
- Takahashi Y, Fukusato T. Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. WJG. 2014;20(42):15539–15548.
- Spruss A, Bergheim I. Dietary fructose and intestinal barrier: potential risk factor in the pathogenesis of nonalcoholic fatty liver disease. J Nutr Biochem. 2009;20(9):657–662.
- Musso G, Gambino R, De Michieli F, et al. Adiponectin gene polymorphisms modulate acute adiponectin response to dietary fat: possible pathogenetic role in NASH. Hepatology 2008;47(4):1167–1177.
- Seki S, Kitada T, Yamada T, et al. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J Hepatol. 2002;37(1):56–62.
- Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Rad Biol Med. 2012;52(1):59–69.
- Serviddio G, Bellanti F, Vendemiale G. Free radical biology for medicine: learning from nonalcoholic fatty liver disease. Free Rad Biol Med. 2013;65:952–968.
- Bellanti F, Villani R, Facciorusso A, et al. Lipid oxidation products in the pathogenesis of non-alcoholic steatohepatitis. Free Radic Biol Med. 2017;111:173–185.
- Liu W, Baker SS, Baker RD, et al. Antioxidant mechanisms in nonalcoholic fatty liver disease. CDT. 2015;16(12):1301–1314.
- Czaja MJ. Induction and regulation of hepatocyte apoptosis by oxidative stress. Antioxid Redox Signal. 2002;4(5):759–767.
- Malhi H, Gores GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28(04):360–369.
- Schreuder TC, Verwer BJ, van Nieuwkerk CM, et al. Nonalcoholic fatty liver disease: an overview of current insights in pathogenesis, diagnosis and treatment. WJG. 2008;14(16):2474–2486.
- Bekyarova G, Tzaneva M, Bratoeva K, et al. Heme- oxygenase-1 upregulated by S-adenosylmethionine potential protection against non-alcoholic fatty liver induced by high fructose diet. Farmatia 2017;65(2):262–267.
- Mantena SK, Vaughn DP, Andringa KK, et al. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem J. 2009;417(1):183–193.
- Simões ICM, Fontes A, Pinton P, et al. Mitochondria in non-alcoholic fatty liver disease. Int J Biochem Cell Biol. 2018;95:93–99.
- Sundaram SS, Halbower A, Pan Z, et al. Nocturnal hypoxia-induced oxidative stress promotes progression of pediatric non-alcoholic fatty liver disease. J Hepatol. 2016;65(3):560–569.
- Videla LA, Rodrigo R, Araya J, et al. Oxidative stress and depletion of hepatic long-chain polyunsaturated fatty acids may contribute to nonalcoholic fatty liver disease. Free Rad Biol Med. 2004;37(9):1499–1507.
- Yesilova Z, Yaman H, Oktenli C, et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic fatty liver disease. Am J Gastroenterol. 2005;100(4):850–855.
- Baskol G, Baskol M, Kocer D. Oxidative stress and antioxidant defenses in serum of patients with non-alcoholic steatohepatitis. Clin Biochem. 2007;40(11):776–780.
- Younossi ZM, Baranova A, Ziegler K, et al. A genomic and proteomic study of the spectrum of nonalcoholic fatty liver disease. Hepatology 2005;42(3):665–674.
- Fu A, Shi X, Zhang H, et al. Mitotherapy for fatty liver by intravenous administration of exogenous mitochondria in male mice. Front Pharmacol. 2017;8:241
- Caldwell SH, Swerdlow RH, Khan EM, et al. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J Hepatol. 1999;31(3):430–434.
- Li S, Takahara T, Fujino M, et al. Astaxanthin prevents ischemia-reperfusion injury of the steatotic liver in mice. PLoS ONE. 2017;12(11):e0187810.
- Voehringer DW, Meyn RE. Redox aspects of Bcl-2 function. Antiox Redox Signal. 2000;2(3):537–550.
- Martínez-Chantar ML, García-Trevijano ER, Latasa MU, et al. Importance of a deficiency in S-adenosyl-L-methionine synthesis in the pathogenesis of liver injury. Am J Clin Nutr. 2002;76(5):1177S–1182S.
- Garcia-Ruiz C, Fernandez-Checa JC. Mitochondrial glutathione: hepatocellular survival-death switch. J Gastroenterol Hepatol. 2006;21:S3–S6.
- Ribeiro PS, Cortez-Pinto H, Solá S, et al. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol. 2004;99(9):1708–1717.
- Ding WX, Yin XM. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J Cell Mol Med. 2004;8(4):445–454.
- Singh R, Wang Y, Schattenberg JM, et al. Chronic oxidative stress sensitizes hepatocytes to death from 4-hydroxynonenal by JNK/c-Jun overactivation. Am J Physiol Gastrointest Liver Physiol. 2009;297(5):G907–G917.
- Ibrahim MA, Kelleni M, Geddawy A. Nonalcoholic fatty liver disease: current and potential therapies. Life Sci. 2013;92(2):114–118.
- Tsuruta F, Sunayama J, Mori Y, et al. JNK promotes bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 2004;23(8):1889–1899.
- Malhi H, Bronk SF, Werneburg NW, et al. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281(17):12093–12101.
- Jeong HS, Kim KH, Lee IS, et al. Ginkgolide A ameliorates non-alcoholic fatty liver diseases on high fat diet mice. Biomed Pharmacother. 2017;88:625–634.