
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Post-transcriptional gene silencing is an effective tool for viral replication control at the epigenetic level. In this study, we aimed to produce dsRNAs and siRNA pools targeting the genetic region for the viral protease 3C and the genetic region covering the sequences for VP1 and VP3 capsid proteins of the Coxsackievirus B3 cardiotropic Woodruff strain by the use of the polymerase complex of bacteriophage Phi6 in HEp-2 monolayer cells. The dsRNAs obtained in this study, specific for the 3C and VP1-VP3 genetic regions of the virus, were characterized by very low cytotoxicity (up to 200 nmol/L and 100 nmol/L, respectively) and high efficiency at silencing the target genes and blocking the enteroviral replication in vitro (93% and 60% reduction of the virus infected cells, respectively, at concentrations of 40 nmol/L). The generated pools of overlapping siRNAs targeting the 3C or VP1-VP3 genetic regions showed insignificant cytotoxicity at concentrations up to 200 nmol/L (< 3%) and higher efficacy than the corresponding dsRNAs. We managed to achieve 95% inhibition of the CVB3 Woodruff strain replication in vitro by using siRNAs specific for the 3C coding region of the virus at a concentration of 40 nmol/L, with no cytotoxicity.
Introduction
Enteroviruses comprise one of the most common groups of human pathogens causing a broad range of diseases. The heaviest clinical manifestations include poliomyelitis and other neurological disorders, encephalitis, neuritis, polyneuritis, meningitis, pericarditis, myocarditis, dilated cardiomyopathy, diabetes and pleurodynia. Coxsackievirus B group, and especially Coxsackievirus B3 (CVB3), includes serotypes related to cardiac infections. In chronic cardiac infections, Coxsackieviruses continue to exist in a latent form [Citation1]. Studies in in vitro and in vivo models conducted over the past 25 years have contributed significantly to elucidate important aspects of the molecular pathogenic mechanisms of cardiac Coxsackievirus infections [Citation2–7]. Based on these studies, it is clear that both directly induced viral lesions and immune and autoimmune mechanisms induced by viral infection are involved in the development of Coxsackievirus induced myocarditis [Citation6–8].
RNA gene silencing is a novel strategy for inhibiting CVB3. Several initial studies achieved variable efficacy with siRNAs targeting the coding regions for 2A [Citation9,Citation10], VP1 [10–12] and 3D RNA-dependent RNA polymerase [Citation10,Citation13]. Gene silencing by siRNAs targeted at the CVB3 plus strand, the minus strand or both simultaneously suggests that gene silencing of the viral plus chain is key to inhibiting CVB3, because the negative strand may be inaccessible to the gene silencing apparatus [Citation14]. Moreover, Schubert et al. [Citation15] found that complex target structures may limit the applicability even of carefully chosen siRNAs. In 2012, the findings of Petrov and Galabov [Citation16] suggested that dsRNAs that are too large cannot influence the CVB1 and CVB3 virus replication in vitro and that the absence of a transfecting agent results in a weak inhibitory effect of dsRNAs. Yuan et al. [Citation10] reported that only siRNAs against the coding region of the viral genome show significant antiviral activity but not against the non-translating regions. Conversely, Petrov and Galabov [Citation17] achieved full inhibition of the CVB3 virus replication by siRNAs targeting the 5'UTR genetic region. Despite these findings, research in this area is still in a preliminary stage, and until now, there is no effective medicine for treatment of the very dangerous CVB viral infections of the brain, nervous system and the heart. In this regard, much more experimental data concerning different viral genetic regions, different strains and different conditions are needed.
In this study, we aimed to produce dsRNAs and siRNAs targeting the genetic region for the viral protease 3C and the genetic region covering the sequences for VP1 and VP3 capsid proteins.
Materials and methods
Materials
Cell cultures
In vitro experiments were performed using monolayer cell cultures of human epithelial cells from a larynx carcinoma (HEp-2). Monolayers were grown in Dulbecco's modified Eagle’s medium (DMEM) containing 5% fetal bovine serum in 96-well plates in a CO2-incubator at 37 °C and 5% CO2.
Virus
Coxsackievirus B3 strain Woodruff (cardiotropic) was obtained from the collection of the Department of Virology, Stephan Angeloff Institute of Microbiology, Bulgarian Academy of Sciences, Sofia, Bulgaria. Infectious titre in HEp-2 cells was 104.5 cell culture infectious dose 50% (CCID50).
Reagents for total RNA extraction
The buffers are listed in .
Table 1. Reagents for extraction of total RNA with silica particles.
Reference compounds
dsRNAs and siRNAs of the S-segment of bacteriophage Phi6 with length of 2948 bp and ∼25 bp, respectively (obtained from the Laboratory of Molecular Virology, University of Helsinki, Finland).
Transfection agent
DharmaFECT1 (Dharmacon, Inc., USA).
Methods
RNA еxtraction
Total RNA was extracted by the use of silica particles [Citation18]. The protocol consisted of two parts: (1) preparation of silica particles and (2) total RNA extraction.
1. Sixty grams silica particles (S5631, Sigma-Aldrich Inc., USA) were mixed with 500 mL ultraclean water. After 24 h the supernatant was removed and another 500 mL of ultraclean water were added. After 5 h the supernatant was removed and the sediment was adjusted to рН 2.0 with HCl. Aliquots of 200 µL were autoclaved at 121°С for 20 min and stored at 4°С until use.
2. Five hundred microliter cell suspension was homogenized with 500 µL Grinding buffer (). Then, 500 µL of the mixture was incubated with 100 µL 10% (w/v) N-Laurilsarcosyl at 70°С for 10 min on a shaker, left for 5 min in ice and centrifuged at 13,000 rpm (MiniSpin, Eppendorf Int.) for 10 min at room temperature (RT). Next, 300 µL of the supernatant was mixed with 150 µL ethanol, 300 µL of 6 mol/L NaJ and 25 µL silica particles and incubated at RT for 10 min min on a shaker. After centrifuging at 6000 rpm (MiniSpin, Eppendorf Int.) for 1 min, the supernatant was removed and the silica sediment was resuspended in 500 µL Washing Buffer (WB) (). This step was repeated twice. The sediment was left to dry at RT, resuspended in 150 µL of ultraclean water and incubated at 70°С for 4 min. After centrifuging at 13,000 rpm (MiniSpin, Eppendorf Int.) for 3 min at RT, 130 µL of the supernatant was collected and stored at –20°С.
Synthesis of DNA template
Touch-down RT-PCR [Citation12] was used for amplification of specific conserved fragments from 3C and VP1-VP3 from CVB3 genome. The primer pairs were designed using CVB3 viral sequences from the National Center for Biotechnology Information (NCBI) database and the BLAST and Clustal Omega tools for alignment (, primers 1–4). Copy DNA was obtained after a denaturation step of 3 μL RNA at 95°С for 5 min with 7 μL of each relevant primer in a final volume of 10 μL and incubation with a master mix containing: 5 μL 5× MMLV-buffer, 2 μL dNTPs (2 mmol/L), 0.5 μL M-MuLV reverse transcriptase (200 U/μL), 7.5 μL DNase and RNase free H2O at 42°С for 60 min. Preparation of the master mix for the following PCR was in a final volume of 25 μL (6 μL cDNA, 2.75 μL 10× PCR buffer, 2.2 μL MgCl2 (25 mmol/L), 2.2 μL dNTPs (2 mmol/L), 1 μL of each primers (10 μmol/L), 1 μL proofreading Pfu DNA-polymerase (5 U/μL) and 8.85 μL H2O) in Auto-Q Server (LKB Instruments Ltd., UK). The touch-down PCR program is given in . The resulting template DNA fragments were separated in 2% agarose gel in ТАЕ buffer with ethidium bromide (0.2 μg/mL) at 100 V for 1 h. PCR products were visualized in trans-illuminator GenoPlex (VWR) with λ = 315 nm.
Table 2. Primers used in touch-down RT-PCR.
Table 3. Program for touch-down RT-PCR.
Synthesis of dsRNA
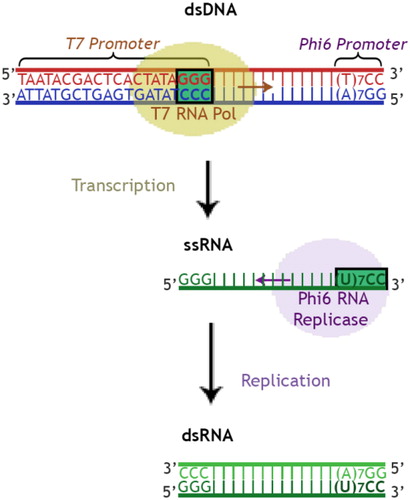
The produced PCR fragments from touch-down RT-PCR served as templates. The primers (, primers 5–8) were designed to include 17–22 nucleotides of the target gene-specific sequence (3C, VP1-VP3) at the 3′-end and RNA polymerase promoter sequences at their 5′ ends so that the resulting PCR products contained the promoter sequences flanking the target sequence ().
Figure 1. Schematic illustration of in vitro dsRNA production system.
Generation of siRNA
We used the recombinant endo-ribonuclease from Giardia intestinalis - Power Cut Dicer to effectively cut dsRNA into a pool of overlapping sequences of siRNAs with a length of approximately 20–30 nucleotides.
Transfection of cells
HEp-2 cells in 96-well plates were transfected with dsRNAs and siRNAs (in concentrations 10, 15, 20, 25, 30, 35, 40, 100 and 200 nmol/L) using DarmaFECT1. The transfection procedure for one well included: (1) preparation of stock solution from each concentration of dsRNAs/siRNAs in 1x siRNA buffer from DarmaFECT1, (2) preparation of two separate tubes with final volumes of 10 µL: 0.5 μL of each concentration dsRNAs/siRNAs with 9.5 μL of DMEM (tube 1) and 0.4 μL DharmaFECT reagent with 9.6 μL of DMEM (tube 2), mixing and incubation at RT for 5 min for each tube, (3) mixing the contents of tube 1 and tube 2 and incubation at RT for 20 min, (4) adding 100 μL transfection medium (20 μL tube mix and 80 μL DMEM) to the HEp-2 monolayer and incubation at 37 °C in 5% CO2 for 24 h.
Determination of cytotoxicity of dsRNAs and siRNA pools
HEp-2 cells were cultured in 96-well plates (Cellstar®, Sigma-Aldrich, USA) and incubated at 37 °C, 5% CO2, for 12 h until formation of confluent monolayer with a density of 2 × 105 cells/mL. Growth DMEM was removed and the transfection procedure (described above) followed. Four replicates per sample were done. Transfection medium was removed and cells were incubated for 60 h and washed with 150 µL/well neutral red (NR) in concentration 0.4% stock solution diluted 1:80 with DMEM. Cells were incubated for another 3 h at 37 °C, 5% CO2 to let the dye penetrate the viable cells. The cells were washed again with 150 µL/well of phosphate buffered saline (PBS) and then, 100 µL/well of extraction solution (1% glacial acetic acid, 50% ethanol and 49% distilled water) was added. The plates were shaken for 10 min and the absorbance of the samples from each well was read on an ELISA Reader (Organon GmbH, Germany) at a wavelength of 540 nm and a reference value of 620 nm. The viability of cells was calculated as a percent from the control (untreated) cells, which were considered as 100% viable, according to the formula:
(1)
(1)
where OP is the optical density (absorption) of live cells.
Determination of the antiviral effect of the dsRNAs and siRNA pools
HEp-2 cells were cultured in 96-well plates (Cellstar®, Sigma-Aldrich, USA) and incubated at 37 °C, 5% CO2, for 12 h until formation of confluent monolayer with a density of 2 × 105 cells/mL. Growth DMEM was removed and the transfection procedure followed. Transfection medium was aspirated from each well and the cells were inoculated with a virus dilution corresponding to a multiplicity of infection (MOI) 0.004. After absorption for 1 h, the unabsorbed virus was removed and the plates were incubated with DMEM for 35 h at 37 °C and 5% CO2. Then, DMEM was removed and cells were washed with 150 µL/well NR (0.4% stock solution diluted 1:80 with DMEM). Plates were incubated for another 3 h at 37 °C and 5% CO2 to let the dye penetrate the viable cells. The cells were washed again with 150 µL/well of PBS (without Ca2+ and Mg2+), the PBS was decanted and treated drop-wise with 100 mL/well of extraction solution (1% glacial acetic acid, 50% ethanol and 49% distilled water). The plates were incubated for 10 min and the absorbance of the samples from each well was read on an ELISA Reader (Organon GmbH, Germany) at a wavelength of 540 nm and a reference value of 620 nm [Citation19]. Trials were performed in four replicates. Controls were non-inoculated cells (health control), virus-inoculated untreated cells (virus control) and non-inoculated treated cells (cytotoxicity control) cultivated under the same conditions. We used referent compounds as internal controls. The antiviral effect was calculated as a percent according to the formula:
(2)
(2)
where OP is the optical density (absorption).
Results and discussion
In this study, we used a method for production of specific dsRNAs and siRNAs targeting selected regions of the viral genome, which we have optimized in previous studies [Citation12,Citation16,Citation17,Citation20].
The first initial DNA template was synthesized by RT-PCR using primers 1 and 2 (), flanking the 3C-RNA genetic region of CVB3 (549 nt). The resulting DNA template fragment of 663 bp, was used to produce specific dsRNAs (of 692 bp) complementary to that region by the use of primers 5 and 6 (). The second initial DNA template was synthesized by RT-PCR using primers 3 and 4 (), flanking the VP3-VP1-RNA genetic region of CVB3 (658 nt). The resulting DNA template of 677 bp, was used to produce specific dsRNAs (of 706 bp) complementary to VP3-VP1 by the use of primers 7 and 8 (). Power Cut Dicer endo-ribonuclease was used to generate pools of siRNAs from the produced target-specific dsRNAs. Both dsRNAs and siRNAs were tested in the following trials.
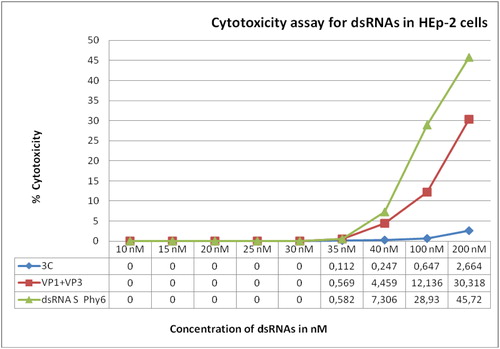
Citotoxicity assay was performed with nine different concentrations from 200 to 10 nmol/L, 200 nmol/L being the undiluted solution of the produced dsRNAs. DsRNAs for the S-segment of bacteriophage Phi6 were used as reference. The highest detected cytotoxicity on the HEp-2 cell monolayer was observed for the dsRNAs for the S-segment of Phi6 at all concentrations from 200 to 35 nmol/L (). Lowest cytotoxicity was observed for the 3C-specific dsRNAs, with highest toxicity values significantly lower (0.2664–0.112%) than the values of VP1-VP3-specific dsRNAs (30.318–0.569%) and S-Phi6-specific dsRNAs (45.72–0.582%). No cytotoxicity was detected for concentrations up to 30 nmol/L ().
Figure 2. Cytotoxicity assay for 3C and VP1 + VP3 specific dsRNAs in HEp-2 cells.
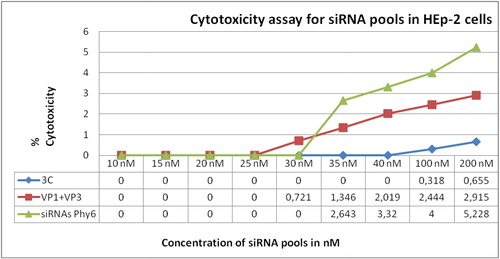
Cytotoxicity assay for siRNAs was conducted with the same concentrations. Similar to the previous experiment, highest cytotoxicity was observed for siRNAs for the S-segment of Phi6 (5.228–2.463% for concentrations 200–35 nmol/L) and lowest, for the 3C-specific siRNAs (<0.66%; ). It should be noted that the cytotoxicity of the produced siRNAs was approximately fourfold, tenfold and ninefold lower than the cytotoxicity of the produced dsRNAs specific for the 3C, VP1-VP3 and S-Phi6 regions, respectively. Furthermore, 3C-specific siRNAs showed no cytotoxicity at concentrations up to 40 nmol/L (). Since the highest cytotoxicity values for both undiluted dsRNAs and siRNAs were below 50%, it was not possible to calculate the half-maximal cytotoxic concentration (CC50) value. Based on the data from the cytotoxicity assays, concentrations of up to 40 nmol/L of the target-specific and referent dsRNAs and siRNAs were applied in the subsequent trials.
Figure 3. Cytotoxicity assay of siRNA pools for HEp-2 cells.
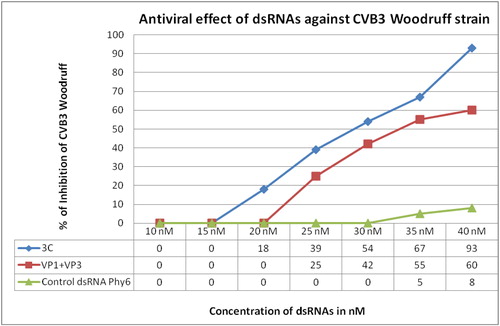
The lowest concentration of dsRNAs with antiviral effect was 20 nmol/L of 3C-specific dsRNAs, which was associated with 18% inhibition of CVB3 titre (). With these dsRNAs we also achieved the highest inhibition of the virus at a concentration of 40 nmol/L – 93%. Referent dsRNAs, specific for the S-segment of Phi6, showed only 8% inhibition of the virus at the highest concentration tested.
Figure 4. Antiviral effect of dsRNAs against Coxsackievirus B3.
An antiviral effect on CVB3 Woodruff strain with the same trend was observed in the assays with siRNAs. Inhibitory effect was observed for 3C-specific siRNAs for a concentration as low as 15 nmol/L (). 3C-specific siRNAs also showed the highest percent of inhibition of the virus (95%) at a concentration of 40 nmol/L. VP1-VP3-specific siRNAs were able to inhibit the viral replication up to 70% at the highest tested concentration. In contrast, the inhibitory effect of the referent siRNAs remained low ().
Figure 5. Antiviral effect of siRNA pools against Coxsackievirus B3.
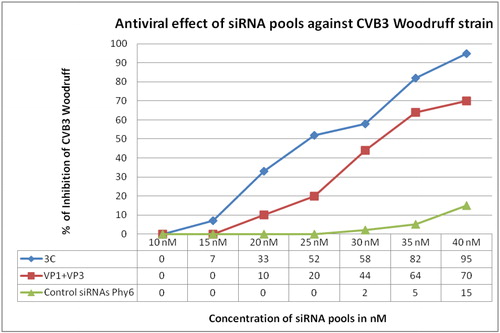
Compared to the dsRNAs specific for 3C and VP1-VP3 genetic regions of CVB3, the application of siRNA pools specific for the same regions, to the CVB3-infected HEp-2 cell monolayer resulted in slightly higher inhibition of the virus. This effect could be observed as: (1) lower concentrations with inhibition of the virus for siRNAs compared to dsRNAs, (2) generally higher inhibition values at the comparative concentrations and (3) higher inhibition values at the highest tested concentrations of dsRNAs and siRNAs ( and ).
Our results are indicative of the excellent potential of siRNAs specific for the 3C genetic region for treatment of infections caused by CVB3 cardiotropic Woodruff strain. Previous studies have shown the potential of siRNAs targeting other viral regions for inhibition of viral replication [Citation9–13,Citation17]. Other studies aimed at silencing of Coxsackievirus adenovirus receptor (CAR) of cardiac myocytes with a variable degree of success [Citation13,Citation21,Citation22] or at modulating the virus-triggered inflammatory response by silencing the colony stimulating factor 1 (CSF-1) in CVB3 infected myeloid cells [Citation23]. 2A, VP1 and 3D RNA-dependent RNA polymerase have been the most exploited gene regions for gene silencing of CVB genes [Citation9–13,Citation17]. Later investigations revealed the great potential of noncoding regions like 5’UTR of CVB3 to be used as alternative targets for specific siRNAs [Citation17]. Silencing of the nontranslated region si5U in another Coxsackievirus, Coxsackievirus A24, also efficiently stopped the virus propagation in vitro [Citation24]. The 3C genetic region of CVB3 is another promising target for post-transcriptional gene silencing and virus control.
Conclusions
The obtained dsRNAs specific for the 3C or VP1-VP3 genetic regions of the virus were produced by using the polymerase complex of bacteriophage Phi6. They showed very low cytotoxicity and high efficiency at silencing the target genes and blocking the enteroviral replication in vitro. The generated pools of overlapping siRNAs showed even lower cytotoxicity and higher efficacy than the corresponding dsRNAs. We managed to achieve 95% inhibition of the CVB3 Woodruff strain replication in vitro by using siRNAs specific for the 3C coding region of the virus.
| Abbreviations | ||
| PTGS | = | post-transcriptional gene silencing |
| dsRNAs | = | double-stranded RNAs |
| siRNAs | = | small interfering RNAs |
| CVB3 | = | Coxsackievirus В3 |
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Tam PE, Messner RP. Molecular mechanisms of Coxsackievirus persistence in chronic inflammatory myopathy: viral RNA persists through formation of a double-stranded complex without associated genomic mutations or evolution. J Virol. 1999;73(12):10113–10121.
- Li-Sha G, Li L, De-Pu Z, et al. Ivabradine treatment reduces cardiomyocyte apoptosis in a murine model of chronic viral myocarditis. Front Pharmacol. 2018;9:182. doi:10.3389/fphar.2018.00182
- Jiang D, Li M, Yu Y, et al. microRNA-34a aggravates Coxsackievirus B3-induced apoptosis of cardiomyocytes through the SIRT1-p53 pathway. J Med Virol. 2019;91(9):1643–1651. doi:10.1002/jmv.25482
- Peischard S, Ho HT, Theiss C, et al. A kidnapping story: how Coxsackievirus B3 and its host cell interact. Cell Physiol Biochem. 2019;53(1):121–140. doi:10.33594/000000125
- Zhang Z, Long C, Li X, et al. CEACAM-1 promotes myocardial injury following Coxsackievirus infection by regulating the Coxsackievirus-adenovirus receptor. Medicine (Baltimore). 2019;98(19):e15629. doi:10.1097%2FMD.0000000000015629
- McManus BM, Chow LH, Wilson JE, et al. Direct myocardial injury by enterovirus: a central role in the evolution of murine myocarditis. Clin Immunol Immunopathol. 1993;68(2):159–169.
- Huber SA. Coxsackievirus-induced myocarditis is dependent on distinct immunopathogenic responses in different strains of mice. Lab Invest. 1997;76(5):691–701.
- Pappritz K, Savvatis K, Miteva K, et al. Immunomodulation by adoptive regulatory T-cell transfer improves Coxsackievirus B3-induced myocarditis. FASEB J. 2018;32(11):6066–6078.
- Merl S, Michaelis C, Jaschke B, et al. Targeting 2A protease by RNA interference attenuates coxsackieviral cytopathogenicity and promotes survival in highly susceptible mice. Circulation. 2005;111(13):1583–1592.
- Yuan J, Cheung PK, Zhang HM, et al. Inhibition of Coxsackievirus B3 replication by small interfering RNAs requires perfect sequence match in the central region of the viral positive strand. J Virol. 2005;79(4):2151–2159.
- Ahn J, Jun ES, Lee HS, Yoon SY, Kim D, et al. A small interfering RNA targeting Coxsackievirus B3 protects permissive HeLa cells from viral challenge. J Virol. 2005;79(13):8620–8624.
- Petrov N, Galabov A. Influence of specific siRNAs on the replication of Coxsackieviruses. In: Galabov AS, Angelova M, Najdenski H, editors. Proceedings of the Third congress of Virology (Days of virology in Bulgaria) with international participation; 2012 Oct 25–27; Sofia, Bulgaria. Sofia: Press Product Line; 2012. p. 21–25.
- Werk D, Schubert S, Lindig V, et al. Developing an effective RNA interference strategy against a plus-strand RNA virus: silencing of Coxsackievirus B3 and its cognate Coxsackievirus-adenovirus receptor. Biol Chem. 2005;386(9):857–863.
- Schubert S, Rothe D, Werk D, et al. Strand-specific silencing of a picornavirus by RNA interference: evidence for the superiority of plus-strand specific siRNAs. Antivir Res. 2007;73(3):197–205.
- Schubert S, Grunweller A, Erdmann VA, et al. Local RNA target structure influences siRNA efficacy: systematic analysis of intentionally designed binding regions. J Mol Biol. 2005;348(4):883–893.
- Petrov N, Galabov A. Conserved regions from Coxsackievirus genome as targets for gene silencing (RNAi). Sci Technol. 2012;II(1):32–36.
- Petrov N, Galabov A. Small Interfering RNAs against human enteroviral infections. Acta Microbiol Bulg. 2016;32(2):101–107.
- Rott ME, Jelkmann W. Characterization and detection of several filamentous viruses of cherry: adaptation of an alternative cloning method (DOP-PCR) and modification of an RNA extraction protocol. Eur J Plant Pathol. 2001;107(4):411–420.
- Borenfreund E, Puerner JA. A simple quantitative procedure using monolayer cultures for cytotoxicity assays (HTD/NR-90). Methods Cell Sci. 1985;9:7–9.
- Petrov N, Bamford D, Galabov A. Production of dsRNAs for induction of posttranscriptional gene silencing against Coxsackievirus B1 infection. Sci Technol. 2011;1(1):255–258.
- Fechner H, Pinkert S, Wang X, Sipo I, et al. Coxsackievirus B3 and adenovirus infections of cardiac cells are efficiently inhibited by vector-mediated RNA interference targeting their common receptor. Gene Ther. 2007;14(12):960–971.
- Sharma M, Mishra B, Saikia UN, et al. Inhibition of Coxsackievirus infection in cardiomyocytes by small dsRNA targeting its cognate Coxsackievirus adenovirus receptor. Indian J Med Res. 2017;146(4):520–527.
- Meyer IS, Goetzke CC, Kespohl M, et al. Silencing the CSF-1 axis using nanoparticle encapsulated siRNA mitigates viral and autoimmune myocarditis. Front Immunol. 2018;9 2303. doi:10.3389/fimmu.2018.02303
- Mishra AK, Satpathy G. Inhibition of Coxsackievirus A24 in permissive Hep2 cells by small interfering RNA (siRNA). IOSRJPBS. 2017;12(03):21–26.