
Abstract
Phloroglucinol (PG) is an important chemical that has been produced in engineered E. coli. Low yield and high cost are the bottlenecks that limit the industrialisation and popularisation of PG biosynthesis. In this study, a new expression system (pNEW vector) was developed for PG biosynthesis by using inexpensive p-isopropylbenzoate (cumate) as the inducer, and the copy number control element (rop) was added to optimise the original pNEW vector, which not only promoted the PG yield, but also reduced the leakage expression. Compared to traditional IPTG inducible expression system, 2-fold enhancement of PG yield was obtained with optimised expression system and the cost of inducer was reduced to 1/9. These results provide technical support for the biosynthesis of PG and reference for the selection of induced expression systems for other chemical biosynthesis.
Introduction
Phloroglucinol (PG) is a very important bulk chemical that is a versatile precursor for the production of pharmaceuticals, food additives and cosmetics, as well as a useful chemical in replication and textile dyeing processes [Citation1]. Therefore, there is a large demand at home and abroad. Current methods for synthesising PG include chemosynthesis and biosynthesis. Owing to lagging in production technology, the traditional chemosynthesis causes serious environmental pollution easily. Besides, the quality of chemosynthetic products cannot keep up with the development of the pharmaceutical industry [Citation2]. However, biosynthesis of PG possesses many advantages inaspects of cost and environment, which has become the development direction of PG biosynthesis [Citation3, Citation4].
At present, many important advances have been made in the biosynthesis of PG. PG can be produced by expressing the phlD gene (encoding Phloroglucinol synthase) derived from Pseudomonas fluorescens (P. fluorescens) in E. coli BL21 (DE3)[Citation5]. The genes used for the formation of acetate and ethanol in E. coli BL21 (DE3) were knocked out; meanwhile, isopropyl-ß-D-thiogalactopyranoside (IPTG)-induced pRSFDuet-1 vector or pACYCDuet-1 vector were used to overexpress acetyl-CoA carboxylase (Accase) from Corynebacterium glutamicum and acetyl-CoA synthetase (Acs) from Escherichia coli (E. coli), respectively, to promote malonyl-CoA, thereby increasing the yield of PG [Citation6]. Then, the marA gene (multiple antibiotic resistance) and Accase were overexpressed by using IPTG-induced pET30a vector or pACYCDuet-1 vector to increase the yield of PG [Citation7]. IPTG-induced pET28a or pACYCDuet-1 as a vector was used to achieve directed evolution of the phlD gene, which increased the thermostability of the PhlD and PG yields compared to the wild type. These researches show that IPTG was used as common inducer to overexpress gene regarding PG biosynthesis in most reports. Due to the high price of IPTG, the costs of production of PG will be increased, thereby limiting the industrialisation and popularisation of PG. In addition, PG yield is still low at present and difficult to meet the requirements of industrial production. So the production of PG still needs to be further improved.
The cumate (4-isopropylbenzoic acid)-inducible expression system, pNEW vector invented by Choi et al [Citation8], is an emerging induction system with the advantages of lower price and less dosage, which has attracted the attention of researchers [Citation8]. Kim et al. [Citation9] used cumate and IPTG to induce overexpression of genomic and foreign genes in E. coli, respectively, and to synthesise short- and medium-chain alpha, beta-unsaturated carboxylic acids by adjusting the ratio of the two inducers [Citation9]. There are also many researchers who have modified the cumate system to work in Alphaproteobacteria [Citation10], Bacillus [Citation11] or yeasts [Citation12]. However, there are still some problems with the cumate induction system, such as more severe leakage expression than the IPTG-induced expression system. At present, there are few studies on the application of cumate in chemical biosynthesis, which also limits the popularisation of cumate. In this study, cumate was first used for PG synthesis, and the original pNEW expression vector was optimised by adding copy number control elements, which not only effectively improved PG yield and reduced costs of production, but also reduced the leakage expression of the original pNEW expression system. The related results provide technical support for the biosynthesis of PG, and also provide reference for the expression systemsselection for other chemical biosynthesis.
Materials and methods
Bacterial strains, plasmids, and culture media
E. coli DH5α was used for all plasmid constructions. DH5α, BL21 (DE3), JM109 and Top10 were used for expression of recombinant proteins. DH5α, BL21 (DE3)、JM109and Top10 have been deposited in the World Data Centre for Microorgannisms (IAL 2069, UPMC 17, UPMC 19, UPMC 18). The expression vectors pET28a-phlD1 is from our laboratory. The phlD1 gene was cloned into pET28a vector between SalI and NcoI sites. E. coli strains were cultivated in liquid Luria-Bertani (LB) media or on LB agar plates for DNA manipulation. For PG production, recombinant strains were cultured using M9 minimal salts medium supplemented with 1 mM MgSO4, 2.5 g/L glucose and trace element solution. M9 minimal salts medium, containing 9.8 g/L K2HPO4·3H2O, 2.1 g/L citric acid, 3 g/L (NH4)2SO4, 0.3 g/L ammonium ferric citrate, was used for shake flask culture [Citation7]. 50 μg/mL of kanamycinor 50 μg/mL of ampicillin was added to the culture medium if necessary. Cumate was purchased from Shanghai Aladdin Bio-Chem Technology Co., Ltd. (Shanghai, China) and dissolved in 100% ethanol to result in a 1 M stock solution [Citation10]. The dilutions with different concentrations were prepared in ethanol to give a diverse-fold stock. For “no cumate” controls, an equal volume of ethanol was added to cultures. Fast Pure Cell/Tissue Total RNA Isolation Kit was purchased from Vazyme Biotech Co., Ltd (Nanjing, China).
Plasmids construction
The bacterial strains, recombinant plasmids and oligonucleotide primers used in this study are listed in . The operon of pNEW—OpNEW, which included its repressor (cymR) and its promoter, a T5 promoter and a control region CuO, was synthesised by Beijing Genomics Institute (BGI, Beijing, China) and ligated to the pUC57 vector to form a pUC57-OpNEW. Since pET28a differs from pNEW only in the operon, a seamless cloning method was used to link the OpNEW and the non-operated region of the recombinant plasmid pET28a-phlD1 (named noOpET28a-phlD1) to form pNEW-phlD1. The rop gene is a component of pET28a, whose role is to control plasmid copy number [Citation13]. In this study, pNEW-phlD1 with rop and pNEW-phlD1 without rop were constructed, named pNEW-phlD1-rop and pNEW-phlD1-norop, respectively. First, the PCR was performed using pUC57-OpNEW as template to amplify the OpNEW. Then, PCR was performed using pET28a-phlD1 as template to amplify the noOpET28a-phlD1 with rop or noOpET28a-phlD1 without rop. Then, pNEW-phlD1-rop and pNEW-phlD1-norop were formed by the homologous arms of OpNEW and noOpET28a-phlD1 with rop or not in the presence of recombinase.
Table 1. Bacterial strains, plasmids and oligonucleotide primers used in this study.
Protein expression and gel electrophoresis analysis
Single colonies of E. coli strain harboring diverse recombinant plasmids were inoculated into liquid LB broth containing 50 μg/mL of kanamycin and grown overnight. The colonised culture was diluted 1:100 in fresh LB media and incubated under the same conditions. When the cell density (OD) at 600 nm reached about 0.6, IPTG or cumate was added to a final concentration of 0.2 mM, and growth was continued for 3 h or 18 h. The volume of the collected bacteria was calculated based on the measured cell density, so that the amount of the cells was kept consistent. The cells pelleted from culture were dissolved in 4 mL PBS buffer and then disrupted under high pressure. The suspension was centrifuged at 13,000 g for 10 min at 4 °C. The supernatant was retained, heated to 100 °C for 10 min and then analysed by SDS-polyacrylamide gel electrophoresis (PAGE).
Shake flask culture and detection of PG production
Shake-flask experiments were carried out in triplicate series of 250 mL Erlenmeyer flasks containing 50 mL of fermentation media. The culture broth was inoculated with E. coli strains harboring different recombinant plasmids and incubated in a gyratory shaker incubator at 37 °C and 200 rpm. When the OD600nm of the culture reached 0.6-0.8, IPTG or cumate were added to a final concentration of 0.2 mM to induce recombinant protein expression, and the shake flask was transferred to a 30 °C shaker. It has been reported in the literature that PG accumulation hardly increased after 12 h cultivation, so cell density and PG production were measured after 3, 6 and 18 h cultivation [Citation7].
Cell density was measured by spectrophotometer at 600 nm. 5 μL of 0.1, 0.2, 0.5, 0.8, 1.0 and 1.5 g/L PG standards were mixed with 1 mL of 100 mg/L cinnamaldehyde coloring solution and allowed to put at room temperature for 15 min. The absorbance at 446 nm was then measured with a spectrophotometer to draw a standard curve [Citation14]. 1 mL of 100 mg/L cinnamaldehyde coloring solution was mixed with 5 μL fermentation supernatant and put for 15 min at room temperature [Citation15]. The absorbance at 446 nm was then measured by a spectrophotometer. The PG yield was calculated from the standard curve [Citation14]. In addition, the PG production was also detected by high performance liquid chromatography (HPLC). The column was Eclipse Plus C18 (250 × 4.6 nm, 5 μm), the mobile phase was acetonitrile:ddH2O (1:1), the flow rate was 0.25 mL/min, the detection wavelength was 230 nm and the column temperature was 30 °C.
Detection of PG yield under different cumate concentration
Shake-flask experiments were carried out in triplicate series of 250 mL Erlenmeyer flasks containing 50 mL of fermentation media. The culture broth was inoculated with BL21(DE3)/pNEW-phlD1-rop and incubated in a gyratory shaker incubator at 37 °C and 200 rpm. When the OD600nm of the culture reached 0.6-0.8, the 10, 20, 30, 50, 100 or 200 μM cumate was added to the media, respectively. Cell density and PG production were measured after 3, 6 and 18 h cultivation. To ensure accurate results, we calculated the PG yield at the same cell density.
Real-time PCR
Total mRNA was extracted from the samples induced by IPTG or cumate using FastPure Cell/Tissue Total RNA Isolation Kit (Vazyme Biotech Co., Ltd, Nanjing, China), then reverse transcribed into cDNA. The RNA extraction kit and reverse transcription kit were from Vazyme Biotech Co., Ltd (Nanjing, China). Real-time PCR amplification was performed using fully automatic real-time PCR system LightCyclerR480 (US) [Citation16]. The 20 μL mixtures involved in the PCR reaction contained the following components: 10 μL of 2 × ChamQ Universal SYBR qPCR Master Mix from Vazyme (Nanjing, China), 0.4 μL forward primer, 0.4 μL reverse primers, 2 μL reverse transcribed cDNA and 7.2 μL double distilled water. PCR amplification was performed using the initial heat for denaturation at 95 °C for 30 s, followed by 40 cycles at 95 °C for 10 s and 60 °C for 20 s, 1 cycle at 95 °C for 15 s, 55 °C for 60 s and 95 °C for 15 s. Then, melting curve analysis was included at the end of last program. The negative control of deionised water that included no-template control with all sets of responses was used to monitor any possible contamination assay. The threshold cycle (Ct) and melting curves were essential parameters in real-time PCR to assist in data analysis. gapA gene (encoding glyceraldehyde 3-phosphate dehydrogenase) was used as an internal reference gene and the 2-ΔΔCT method was used to analyse the relative mRNA expressions [Citation17]. Primers for real-time PCR are listed in .
Accession number
The gene sequences are available at the GenBank database under accession numbers EU554263.1 (phlD1) and BBH63260.1 (rop).
Results and discussion
Effect of new vector on PG synthesis
A type III polyketide synthase cloned from Pseudomonas fluorescens (encoded by phlD) has been proved to be the key biosynthetic gene for PG [Citation18]. The PG can be produced by heterogolously overexpressing the phlD gene only in E. coli. So the phlD gene was used to study the induction system in this study. The pNEW vector was selected as an expression vector for expressing the phlD1 gene to study the effect of cumate inducer on the expression of phlD1 and PG production. Then, the pNEW plasmid reported in the literature has no rop gene. Therefore, we added a copy number rop gene to the pNEW vector to study the effect of rop on soluble protein expression and PG yields. Recombinant plasmids pNEW-phlD1-ropand pNEW-phlD1-norop were constructed using seamless clone methods. The recombinant plasmid pET28a-phlD1was from our laboratory as control [Citation7]. To verify the difference in expression levels of three plasmids induced by IPTGor cumate, we detected the PG yield, soluble protein expression and mRNA expression levels, respectively.
Effect of cumate-inducible vector on PG yield
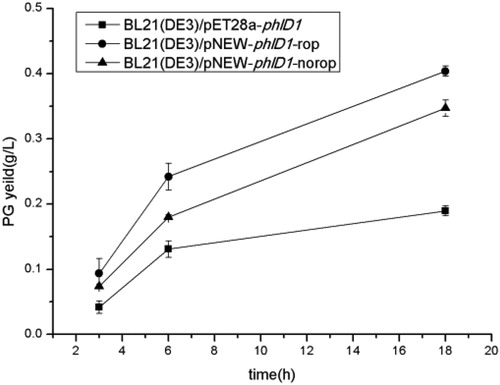
In this study, we examined the effects of three strains on PG biosynthesis by comparing PG yield in the fermentation supernatant (). After shake flask verification, three strains had no significant difference in PG yield after 3 h cultivation. However, the PG yield of BL21(DE3)/pNEW-phlD1-ropreached 0.4 g/L after 18 h cultivation, about 2.2 times higher compared to BL21(DE3)/pET28a-phlD1(0.18 g/L). Moreover, the PG yield of BL21(DE3)/pNEW-phlD1-norop is 15% lower than that of BL21(DE3)/pNEW-phlD1-rop. At the same time, the PG yield was detected by HPLC, and the result was the same as that of cinnamaldehyde colorimetry method. From the above results, it can be seen that the cumate-induced pNEW vector is more suitable for PG production than the IPTG-induced pET28a vector.
Figure 1. PG yields of different strains.
Note: All experiments were done in triplet, and error bars are the standard error of the mean.
Effect of new vector on phlD1 soluble protein
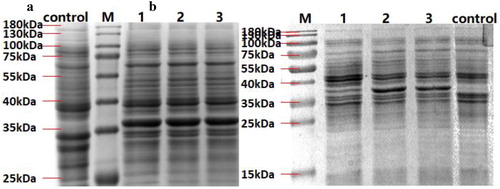
The molecular weight of the PhlD protein is approximately 38 kDa. After 3 h cultivation, the expression of soluble PhlD1 protein in the three strains was not significantly different (), but the difference was significant after 18 h cultivation (). The percentages of bands of soluble PhlD1 protein to total soluble protein were calculated by software Image Lab, which were 2.8%, 14.3% and 11.3%, respectively. It can be seen from the experimental results that the soluble protein of BL21(DE3)/pET28a-phlD1 is much less than that of BL21(DE3)/pNEW-phlD1-rop, and soluble PhlD1 protein of BL21(DE3)/pNEW-phlD1-norop is slightly less than BL21(DE3)/pNEW-phlD1-rop. As we expected, adding rop gene to pNEW vector can increase expression of soluble PhlD1 protein. The trend of the difference in soluble protein of the three strains was consistent with the trend of the difference in PG yield.
Figure 2. The soluble protein of different strains from samples induced after 3 h (a) and 18 h (b).
Note: M: Marker; control: BL21(DE3); 1: BL21(DE3)/pET28a-phlD1; 2: BL21(DE3)/pNEW-phlD1-rop; 3: BL21(DE3)/pNEW-phlD1-norop. The position indicated by the black arrow is the location of PhlD protein.
Effect of new vector on mRNA expression
In order to prove that the phlD1 genes of the three strains are normally expressed and to check whether the expression levels of phlD1 mRNA of the three strains are consistent with the expression trend of soluble phlD1 proteins, real-time PCR was performed.
The results on the graph showed the difference in expression of phlD1 mRNA after 3 () or 18 h () cultivation in three strains. The relative mRNA level of BL21(DE3)/pET28a-phlD1 was more than that of BL21(DE3)/pNEW-phlD1-rop. Meanwhile, the relative mRNA level of BL21(DE3)/pNEW-phlD1-norop was more than that of BL21(DE3)/pNEW-phlD1-rop. Since the T7 promoter is stronger than the T5 promoter and the copy number of the rop-free pNEW vector is more than the copy number of the pNEW vector containing rop, the difference in phlD1 mRNA level in the experimental results is consistent with the theory [Citation13, Citation19]. However, the difference in mRNA levels in the experimental results is opposite to the soluble protein, that is, the higher the mRNA expression level, the lower the soluble protein. The reason may be that too much mRNA is expressed, resulting in PhlD protein expression being too fast to be properly folded and the formation of insoluble inclusion bodies [Citation20]. As we reported previously, although the Tac promoter is weaker than the T7 promoter, it shows higher PG yield when the Tac promoter is used as the promoter of the gene GroESL that increased the tolerance of E. coli for PG. The reason may be that excessive overexpression of the GroESL protein may cause a metabolic burden on the host strain and weaken its drug resistance [Citation21]. Thus, the results that we reported previously [Citation21] are in line with the results in this study, that is, strong promoters are not necessarily suitable for PG production.
Figure 3. The relative phlD1 mRNA level of different strains from samples induced after 3 h (a) and 18 h (b).
Note: Error bars are the standard error of the mean.
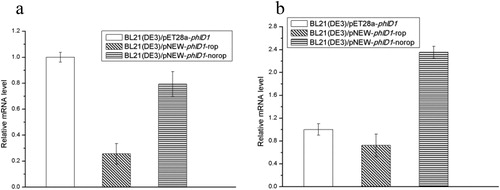
In a word, relative mRNA level expressed by BL21(DE3)/pNEW-phlD1-rop is lower compared to BL21(DE3)/pET28a-phlD1, while the amount of soluble PhlD1 protein is reversed, so the PG yield of BL21(DE3)/pNEW-phlD1-rop is about twice that of BL21(DE3)/pET28a-phlD1. Regarding the pNEW vector with or without rop, BL21(DE3)/pNEW-phlD1-rop has less mRNA than BL21(DE3)/pNEW-phlD1-norop. However, soluble PhlD1 protein expressed by BL21(DE3)/pNEW-phlD1-rop is slightly more compared to BL21(DE3)/pNEW-phlD1-norop, so the PG yield of pNEW-phlD1-norop is 15% lower than that of pNEW-phlD1-rop.
Effect of new vector on leakage expression
We also studied the effect of new expression systems on leakage expression. In the case of no inducer after 3 or 6 h cultivation, the absorbance at a wavelength of 446 nm was smaller than the measurement range of the cinnamaldehyde color developing solution, so the PG yield in the fermentation broth after 18 h cultivation was measured by HPLC. The results showed that the PG yield of BL21(DE3)/pNEW-phlD1-rop was twice that of BL21(DE3)/pET28a-phlD1, but the leakage expression of pNEW-phlD1-rop was also twice that of BL21(DE3)/pET28a-phlD1, up to 0.048 g/L. At the same time, the leakage expression of BL21(DE3)/pNEW-phlD1-norop is more serious than that of BL21(DE3)/pNEW-phlD1-rop, up to 0.074 g/L. For leak expression, the results showed that the leakage expression of BL21(DE3)/pNEW-phlD1-norop is more serious than that of BL21(DE3)/pNEW-phlD1-rop. In the same way as the expression of phlD1, the reason why the pNEW-phlD1-norop leakage expression is more likely is that high-copy plasmid has a high expression level of the CymR protein, resulting in less CymR forming a correct structure, which made the repressive effect worse [Citation22]. At the same time, the leakage expression of pNEW-phlD1-rop was twice that of BL21(DE3)/pET28a-phlD1. The conclusion is that although added the copy number control element (rop) can reduce the leakage expression of pNEW vector, the pNEW vector has more leaky expression than the pET vector. The reason is the repressive effect of the CymR is not rigorous as LacI [Citation23]. Fortunately, other progress had been made to resolve this problem. Hou et al. [Citation23]. increased the stringency of CymR by adding modular oligomerisation domain to the repressor CymR and using a double manipulation region, reducing the leakage expression of pNEW vector by an order of magnitude Therefore, the problem of high leakage expression of the pNEW expression vector can be solved and the pNEW vector will possess larger application potential in the biosynthesis of chemicals such as PG.
Detection of PG production at different cumate concentrations
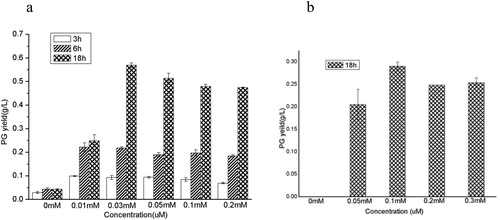
To investigate the optimal concentrations of cumate, phlD gene was induced with different concentrations of cumate. The final concentration of the inducer was 0, 0.01, 0.03, 0.05, 0.1 and 0.2 mM, respectively. The PG yield was detected in shake flask after 3 h, 6 h and 18 h cultivation. It has been reported in the literature that PG accumulation hardly increased after 12 h cultivation, so it is enough to detect PG after 18 h cultivation [Citation7]. As a result, there was no significant difference in PG yield after 3 and 6 h cultivation. Until 18 h cultivation, PG yield under different concentration of cumate was obviously different (). The reason may be that the cell density after 3 or 6 h cultivation was small, and the inducer at the above-mentioned concentrations were excessive for PG production. However, as the cell density increases during 18 h cultivation, when the concentration is 0.1 μM, the inducer can no longer satisfy the PG production, but the concentration of 0.03 μM can. After 18 h cultivation, when the concentration of the inducer was 0.01 mM, the PG yield was about 0.3 g/L, and when the inducer concentration was 0.03 mM, the PG yield was about 0.55 g/L. When the concentration of the inducer increased again, the PG yield did not change. The results showed that the optimal concentration of cumate as an inducer was 0.03 mM. In contrast, the optimal induction concentration of IPTG was 3 times that of cumate, 100 mM. Therefore, the new inducer cumate has an apparent advantage over IPTG. The price of IPTG is 128¥/5 g, while the price of cumate is 48 ¥/5 g, almost 1/3 of IPTG. Moreover, the optimal final concentration of IPTG is 100 μM [Citation7], while cumate is 30 μM, almost 1/3 of IPTG. In other words, the cost of using cumate is 1/9 of IPTG.
Figure 4. PG production under different concentrations of cumate (a) or IPTG (b).
Note: All experiments were done in triplet, and error bars are the standard error of the mean.
The versatility of pNEW vectors in other strains
Since the T5 promoter of the pNEW vector can be recognised by various E. coli, we detected the PG production of pNEW-phlD1-rop in DH5α, JM109and Top10. The results of the shake flask fermentation experiments showed that the recombinant plasmid pNEW-phlD1-rop can produce PG in DH5α, JM109 and Top10, but the PG yield differed in different hosts. After 6 h cultivation, the PG yields of DH5α/pNEW-phlD1-rop and Top10/pNEW-phlD1-rop were not much different, while the PG yield of JM109/pNEW-phlD1-rop was lower, which was 62% of DH5α/pNEW-phlD1-rop or Top10/pNEW-phlD1-rop (). Therefore, the use of cumate-induced pNEW expression vector can expand the range of hosts. Unlike T7 promoter in pET vector, which required the E. coli host strains to be genetically modified in order to express a T7 RNA polymerase for transcription to occur [Citation8], all E. coli capable of recognizing the T5 promoter, such as DH5α, top10 and JM109, can be used as hosts for the pNEW vector.
Table 2. PG yield of pNEW-phlD1-rop in DH5α, JM109 and Top10.
According to recent review, apart from E. coli, Saccharomyces cerevisiae, Streptomyces venezuelae, Corynebacterium glutamicum and Yarrowia lipolytica can all be considered for the PG biosynthesis due to their respective outstanding advantages [Citation24]. Compared to IPTG, cumate is a non-toxic small molecule and low-cost, so it will be more suitable as an inducer for these strains.
Conclusions
In this study, the cumate-inducing system is used for PG biosynthesis. The PG yield of BL21 (DE3)/pNEW-phlD1-rop is twice that of BL21 (DE3)/pET28a-phlD1. Meanwhile, PG yield of BL21 (DE3)/pNEW-phlD1-rop is 15% higher than that of BL21 (DE3)/pNEW-phlD1-norop, while the leakage expression of BL21 (DE3)/pNEW-phlD1-rop is 50% of the leakage expression of BL21 (DE3)/pNEW-phlD1-norop. The gene rop not only increases PG yield, but also reduces leakage expression, making the cumate-inducible expression vector more efficient and rigorous. In addition, the cost of using cumate is 1/9 of IPTG, which greatly reduces the cost of experiments. Finally, the use of cumate-induced pNEW expression vector can expand the range of hosts.
Author contributions
Mo Xian and Wei Liu designed the work. Shan Yuan performed the experiment and wrote the manuscript. Rubing Zhang, Yujin Cao, Jing Guo analysed the data and modified the manuscript.
Compliance with ethical standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Singh IP, Sidana J, Bansal P, et al. Phloroglucinol compounds of therapeutic interest: global patent and technology status. Expert Opin Ther Pat. 2009;19(6):847–866.
- Kastens ML, Kaplan JF. TNT into phloroglucinol. Ind Eng Chem. 1950;42(3):402–413.
- Zhang R, Liu W, Cao Y, et al. An in vitro synthetic biosystem based on acetate for production of phloroglucinol. BMC Biotechnol. 2017;17(1):66.
- Abdel-Ghany SE, Day I, Heuberger AL, et al. Production of phloroglucinol, a platform chemical, in Arabidopsis using a bacterial gene. Sci Rep. 2016;6(1):38483.
- Achkar J, Xian M, Zhao H, et al. Biosynthesis of phloroglucinol. J Am Chem Soc. 2005; 127(15):5332–5333.
- Zha W, Rubin-Pitel SB, Shao Z, et al. Improving cellular malonyl-CoA level in Escherichia coli via metabolic engineering. Metab Eng. 2009;11(3):192–198.
- Cao Y, Jiang X, Zhang R, et al. Improved phloroglucinol production by metabolically engineered Escherichia coli. Appl Microbiol Biotechnol. 2011;91(6):1545–1552.
- Choi YJ, Morel L, Le Franois T, et al. Novel, versatile, and tightly regulated expression system for Escherichia coli strains. Appl Environ Microbiol. 2010;76(15):5058–5066.
- Seohyoung K, Seokjung C, Ramon G. Engineering Escherichia coli for the synthesis of short- and medium-chain α,β-unsaturated carboxylic acids. Metab Eng. 2016;36:90–98.
- Kaczmarczyk A, Vorholt JA, Francez-Charlot A. Cumate-inducible gene expression system for sphingomonads and other Alphaproteobacteria. Appl Environ Microbiol. 2013;79(21):6795–6802.
- Seo SO, Schmidt-Dannert C. Development of a synthetic cumate-inducible gene expression system for Bacillus. Appl Microbiol Biotechnol. 2019;103(1):303–313.
- Shigehito I, Jef DB. New orthogonal transcriptional switches derived from tet repressor homologues for Saccharomyces cerevisiae regulated by 2,4-diacetylphloroglucinol and other ligands. ACS Synth Biol. 2017;6(3):497–506.
- Cesareni G, Helmer-Citterich M, Castagnoli L. Control of ColE1 plasmid replication by antisense RNA. Trends Genet Tig. 1991;7(7):230–235.
- Rao G, Lee JK, Zhao H. Directed evolution of phloroglucinol synthase PhlD with increased; stability for phloroglucinol production. Appl Microbiol Biotechnol. 2013;97(13):5861–5867.
- Zha W, Rubin-Pitel S, Zhao H. Exploiting genetic diversity by directed evolution: molecular breeding of type III polyketide synthases improves productivity. Mol Biosyst. 2008;4(3):246–248.
- Bubner B, Baldwin IT. Use of real-time PCR for determining copy number and zygosity in transgenic plants. Plant Cell Rep. 2004;23(5):263–271.
- Sumby KM, Grbin PR, Jiranek V. Validation of the use of multiple internal control genes, and the application of real-time quantitative PCR, to study esterase gene expression in Oenococcus oeni. Appl Microbiol Biotechnol. 2012;96(4):1039–1047.
- Bangera MG, Thomashow LS. Identification and characterization of a gene cluster for synthesis of the polyketide antibiotic 2,4-diacetylphloroglucinol from Pseudomonas fluorescens Q2-87. J Bacteriol. 1999;181(10):3155–3163.
- Brunner M, Bujard H. Promoter recognition and promoter strength in the Escherichia-Coli system. EMBO J. 1987;6(10):3139–3144.
- Kane JF, Hartley DL. Formation of recombinant protein inclusion-bodies in Escherichia-Coli. Trends Biotechnol. 1988;6(5):95–101.
- Zhang R, Cao Y, Liu W, et al. Improving phloroglucinol tolerance and production in Escherichia coli by GroESL overexpression. Microb Cell Fact. 2017;16(1):227.
- Singh SM, Panda AK. Solubilization and refolding of bacterial inclusion body proteins. J Biosci Bioeng. 2005;99(4):303–310.
- Hou J, Zeng W, Zong Y, et al. Engineering the ultrasensitive transcription factors by fusing a modular oligomerization domain. ACS Synth Biol. 2018;7(5):1188–1194.
- Claire MP, Hal SA. Expanding the chemical palette of industrial microbes: metabolic engineering for type III PKS-derived polyketides. Biotechnol J. 2019;14(1):e1700463.