
Abstract
At present, the oestrogens detected in the environment have been classified as group 1 carcinogens by the World Health Organization. They have obvious effects on organisms at extremely low environmental concentration (1.0 ng/L) and easily accumulate in the ecosystem, which has adverse effects on the environment and ecological health. The use of microbial degradation to remove steroid estrogens from polluted environments has received increasing attention. In this study, a bacterium capable of degrading 17β-estradiol was isolated from a sewage treatment plant in Jilin, China, and identified as Serratia nematodiphila DH-S01. The results of degradation experiments showed that after culturing the bacteria for 4 days, the degradation rate of oestrone and 17β-estradiol at 15 mg/L reached 93.47% and 93.2%, respectively. Genome-wide sequencing showed that the genome of strain DH-S01 consists of a single circular chromosome, 5,256,558 bp in length, which contains 4,874 predicted coding genes. Based on genome annotation, high abundance genes are related to the metabolism of terpenoids and polyketides. Nine types of sterol- and oestrogen-degrading enzymes were annotated in this strain, and the existence and expression of the enzymes were analyzed by polymerase chain reaction (PCR) and reverse transcription polymerase chain reaction (RT-PCR). Comparative genomic analysis showed that there are genes encoding eight enzymes in the common genes of the four Serratia strains, highlighting the potential of the other three Serratia strains to degrade steroid estrogen.
Introduction
Since the beginning of global industrialization, environmental oestrogen levels have developed from an emerging into a severe problem. Worldwide, steroid estrogens pose a severe threat to soil, plants, water resources and humans. In fact, in recent years, the global concentration of steroid estrogens in soil and water has increased rapidly, which causes great concern [Citation1]. Steroid estrogens have high activity and endocrine disruption potential. Even at very low environmental concentrations, they will also have significant effects on organisms (1.0 ng/L) [Citation2], such as disturbing the normal physiological activities, reproduction, development and behaviour of the human body [Citation3] and the reproductive health of aquatic organisms [Citation4,Citation5]. In addition, steroid estrogens tend to accumulate in ecosystems, leading to loss of biological habitats and diversity and a series of adverse effects on the environment and ecological health [Citation6,Citation7]. It is worth noting that the effects of steroid estrogen on the development and function of the reproductive system are not limited to the exposed individuals themselves; in some cases, the offspring is also affected [Citation8]. Oestrogens have been classified as group 1 carcinogens by the World Health Organization (http://monographs.iarc.fr/ENG/Classification/latest_classif.php). With population growth and the development of aquaculture, human and livestock excreta have become the largest source of steroid estrogens in the natural environment, adding natural steroid estrogens to environmental water sources. The resulting pollution is increasingly worsening [Citation9–12].
In addition to physical and chemical methods, microbial biodegradation plays a central role in the removal of steroid estrogens from the natural environment [Citation12,Citation13]. Bacteria capable of oestrogen degradation under aerobic conditions have been isolated and identified from various environments, including activated sludge, compost, soil, sandy aquifers and oceans. These isolated strains include Proteobacteria, Actinobacteria, Bacteroides and Firmicutes [Citation14]. Studies have shown that Proteobacteria was the most dominant community in soil [Citation15], sewage [Citation16] and activated sludge [Citation17]. For example, Fujii et al. [Citation18] reported the isolation and characterization of the first oestrogen-degrading sludge bacterium, Novosphingobium tardaugens ARI-1. Sphingomonas sp. KC8 and Novosphingobium sp. SLCC were used as model microorganisms and their oestrogen aerobic degradation pathways were characterized by oestrogen metabolism profiles and genomic analysis of oestrogen-degrading bacteria [Citation19]. In addition, the carbon atom backbone of a steroid molecule consists of four fused rings (A ring to D ring) and side chains of up to 10 carbon atoms. Key reactions to microbial degradation of steroids include sterol side chain degradation, hydroxylation and redox reactions at different locations in the steroid core [Citation20]. Side chain degradation and steroid core oxidation have been shown to be independent processes, and the order of these processes can vary within one genus [Citation21].
Serratia sp. is a gram-negative bacillus belonging to γ-Proteobacteria and has multiple functions. Serratia proteamaculans, Serratia plymuthica and Serratia marcescens can promote plant growth and inhibit the growth of different plant pathogenic fungi [Citation22–24]. The genomes of several of these strains were sequenced. However, the genome of Serratia sp. that degrades steroid estrogens has not been reported. The whole genome sequence is necessary to describe the degradation mechanism of steroid estrogens in bacteria. In addition, comparative genetic analysis may help to fully understand the functional characteristics of Serratia.
In this study, Serratia nematodiphila DH-S01 was isolated and purified from a sewage treatment plant (STP) in Jilin, China. The strain has the ability to use oestrone and 17β-estradiol as sole carbon source. Genome-wide sequencing and gene function annotation of the strain were performed, enzymes involved in steroid estrogen degradation were predicted and verified by PCR and RT-PCR. In addition, the genomic characteristics of the DH-S01 strain were compared with those of the other three Serratia species. This research laid the foundation for the further exploration of the biochemical mechanisms and molecular mechanisms underlying this steroid estrogen degradation.
Materials and methods
Chemical reagents
The utilized experimental drugs and reagents include oestrone (E1) (purity of 98% produced by Shanghai Jingjing Biochemical Technology Co., Ltd., Shanghai, China), 17β-estradiol (E2) (purity of 98% produced by Shanghai Jingjing Biochemical Technology Co., Ltd., Shanghai, China), methanol (Beijing Chemical Plant, Beijing, China), inorganic salt medium (g/L): 4.26 g Na2HPO4,1.5 g (NH4)2SO4, 2.65 gKH2PO4, 0.2 g MgSO4·7H2O and 0.02 g CaCl2, trace elements (g/L): 0.024 g NiCl2·6H2O, 0.19 g CoCl2·6H2O, 0.002 g CuCl2·2H2O, 0.061 g MnSO4·H2O, 0.024 g Na2MoO4·2H2O, 0.07 g ZnCl2 and 0.006 g H3BO3, Luria-Bertani (LB) medium, phosphate buffer stock solution (pH 7.0), phosphate buffer working solution (pH 7.0), 17β-estradiol selection medium and 17β-estradiol mother liquor.
Isolation and identification of strains
Activated sludge samples were collected from a sewage treatment plant in Jilin Province, China, and were used to enrich and isolate oestrogen-degrading bacteria. The morphological characteristics of single colonies were observed after separation. Then, the strains were picked from single colonies and fixed on slides. After dehydration with different concentrations of ethanol, the strain morphology was observed under a scanning electron microscope (FEl XL-30, USA) [Citation25]. The strain was inoculated in LB liquid medium, cultured in a shaking incubator at 30 °C and 120 rpm for 24 h, and a series of physiological and biochemical experiments were conducted using Enterobacteriaceae bacterial biochemical routine identification kit (Guangdong Huan Kai Microbiology Technology Co., Ltd., Guangzhou, China).
Using universal primers (Supplemental Table S1), the 16S rDNA sequence of the strain was amplified by PCR, and the PCR reaction system is shown in Supplemental Table S2. The PCR product was sequenced using an ABI DNA sequencer (model: 3730XL). The sequencing results were imported into GenBank, and the measured gene sequences were compared with the 16S rDNA sequences in the GenBank database using the BLAST functional component. The 16S rDNA sequences of 18 strains with higher BLAST scores were selected, and phylogenetic tree analysis was performed according to the Neighbor-Joining method using MEGA version 5.2.1 software. The strain has been deposited in the China Center for Type Culture Collection (CCTCC) under accession number CCTCC M 2019038.
Degradation of oestrone and 17β-estradiol by the isolate
The strain was separately inoculated into 100 mL sterile inorganic salt medium (MSM) with 15 mg/L oestrone or 17β-estradiol as sole carbon sources and energy source under optimal conditions (pH = 7.0, 30 °C and 120 rpm). Strains were cultured in triplicate for 6 days. Uninoculated medium was used as control. Samples were periodically taken from the culture, cell densities were measured at OD 600 nm using a microplate reader, and after sample pre-treatment, the remaining concentrations of oestrone or 17β-estradiol in the medium were measured by high performance liquid chromatography (HPLC). For the preparation of standard curves, substrates were prepared at concentrations of 1 mg/L, 5 mg/L, 15 mg/L, 30 mg/L and 50 mg/L. Standard curves were prepared by HPLC. A high-performance liquid chromatograph, manufactured by Shimadzu Corporation, was used with a Zorbax Eclipse Plus C18 column (150 × 4.6 mm, 3.5 mm). The detection conditions were as follows: wavelength 220 nm, acetonitrile/water = 50:50, injection volume 10 μL, flow rate 0.8 mL/min, and room temperature.
Whole genome sequencing and assembly
The strain was inoculated in LB liquid medium, cultured at 30 °C, 120 rpm for 24 h and then centrifuged at 4000 rpm for 10 min. The supernatant was discarded and the cells were resuspended in 0.2 mol/L phosphate buffer (pH 7.0). The cells were again centrifuged and the process was repeated three times to wash the cells. Genomic DNA was extracted using a bacterial genomic DNA extraction kit (Tiangen Biochemical Technology Co., Ltd.). The harvested DNA was detected by agarose gel electrophoresis and quantified with QUBIT 2.0 FLUOROMETER (Invitrogen Co., Ltd., USA). Whole-genome sequencing was performed on the Illumina HiSeq 2500-PE125 platform with massively parallel sequencing (MPS) Illumina technology. A-tailed, ligated to paired-end adaptors and PCR amplified with a 500 bp insert and a mate-pair library with an insert size of 5 kb were used for the library construction. Illumina PCR adapter reads and low-quality reads from the paired-end and mate pair library were filtered by a quality control step using our own compiling pipeline. All good quality paired reads were assembled into a number of scaffolds using SOAP de novo (http://soap.genomics.org.cn/soapdenovo.html) [Citation26,Citation27]. Then, the filtered reads were handled by the next step of the gap-closure. The genomic sequence of S. nematodiphila DH-S01 has been deposited in GenBank under accession number CP038662. The average nucleotide identity (ANI) was calculated using JSpecies software, version 1.2.1 [Citation28].
Whole genome gene annotation
The GeneMarkS program was used to retrieve the related coding gene. Transfer RNA (tRNA) genes were predicted by tRNAscan-SE. Ribosome RNA (rRNA) genes were analyzed by rRNAmmer. Small nuclear RNAs (snRNA) were predicted by BLAST against the Rfam database. The IslandPath-DIOMB program was used to predict Genomics Islands. Seven databases were used to predict gene functions. These were Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Orthologous Groups (COG), Non-Redundant Protein Database (NR), Transporter Classification Database (TCDB), Swiss-Prot and Carbohydrate-Active enZYmes Database (CAZy). A whole genome Blast search (E-value less than 1e-5, minimal alignment length percentage exceeding 40%) was performed against the seven above-mentioned databases.
PCR and RT-PCR detection of steroid estrogen-degrading enzymes
Amplification was performed using reagents from Beijing Quanjin Biotechnology Co., Ltd., including EasyTaq® DNA Polymerase (Cat. No. AP111-03) and High Pure dNTPs (Cat. No. AD101-02). Using the primer design software Primer version 5, primers were designed (). The PCR mixture is shown in Supplemental Table S3. The PCR amplification conditions are as follows: 95 °C for 5 min, 35 cycles of 95 °C for 15 s, 55 °C for 15 s and 72 °C for 3 min, and a final extension at 72 °C for 7 min. The PCR products were subjected to agarose gel electrophoresis. Finally, sequencing was performed using the corresponding amplification primers.
Table 1. PCR primer sequences.
The strain was inoculated into an inorganic salt medium containing 15 mg/L oestrone or 17β-estradiol as sole carbon source, and the control group received sodium acetate as sole carbon source. After culture for 3 days at 30 °C and 120 rpm, the total RNA was extracted using a bacterial total RNA extraction kit (Bacterial RNA Kit; Haoran Biotech). The extracted RNA was then converted to cDNA by using an RT-PCR kit (Dalian Bao Bioengineering Co.), and the obtained cDNA was used as PCR template. The PCR product was analysed by agarose gel electrophoresis and the amplified fragment was detected. Fragments were recovered from the gel and sequenced.
Comparative genomic analysis of Serratia nematodiphila DH-S01
The genome sequences of S. nematodiphila DH-S01 and S. nematodiphila DZ0503SBS1 (GenBank accession number NZ_JPUX00000000.1), Serratia fonticola DSM 4576 (GenBank accession number. NZ_CP011254.1) and S. marcescens Db11 (GenBank accession number NZ_HG326223.1) were compared. DZ0503SBS1, DSM 4576 and Db11 were used as reference genomes. The genomes of target bacteria and reference strains were analyzed simultaneously at the nucleic acid level using MUMmer [Citation29]. The complete genomic sequences of these four Serratia strains (DH-S01, DZ0503SBS1, DSM 4576 and Db11) were aligned and displayed in progressive mode using MAUVE [Citation30].
Results and discussion
Isolation and identification of the strain
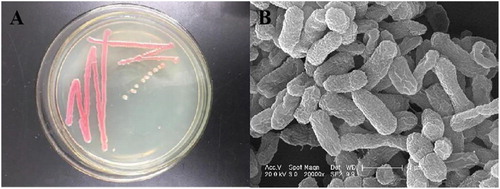
A bacterial strain was isolated from a STP in Jilin, China and was named DH-S01. The strain formed round colonies, the edges are neat, shiny, orange-red or purple-red, the bacteria are easy to provoke (). Electron micrographs of the strains showed that the cells were short rod-shaped (). The physiological and biochemical characteristics of the strain showed identified it as a gram-negative bacterium (Supplemental Table S4). Phylogenetic tree analysis was based on the 16S rDNA sequence of the strain, which has the closest genetic distance to the genus Serratia (Supplemental Figure S1). Based on the physiological and biochemical assays and bacterial morphology, the genus of the strain was identified as Serratia.
Figure 1. Morphological observation of the steroid estrogen-degrading strain DH-S01. Colony morphology (A) and scanning electron micrographs (B).
Oestrone and 17β-estradiol degradation by S. nematodiphila DH-S01
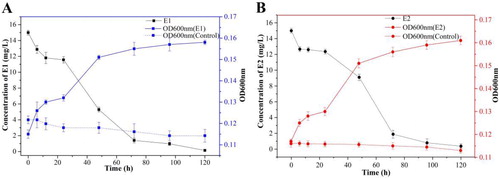
With E1 and E2 as the sole carbon source, respectively, growth and degradation curves of the strains were measured during the culture period ( and B). The strain DH-S01 degraded E1 and E2 during the growth retardation period within the first 24 h of the culture period, which may be caused by the strain’s inability to adapt to the substrate during the early degradation period. After 24 h of cultivation, the strain DH-S01 was adapted and adjusted, and it began to use E1 and E2 as carbon and energy sources for growth and metabolism. The strain DH-S01 entered the logarithmic growth phase, and the OD600 of the bacterial suspension showed an upward trend. At this time, the strain DH-S01 needs to consume a considerable amount of nutrients for growth and metabolism, and the substrate concentration gradually decreased. When cultured to 96 h, the degradation rate of E1 reached 93.47%, and the OD600 value at this time was 0.158. The degradation rate of E2 reached 93.2%, and the OD600 value was 0.160. The results showed that the strain DH-S01 was able to grow with E1 and E2 as the sole carbon source and achieved a significant degradation effect.
Figure 2. Growth of strain DH-S01 and degradation of oestrone (E1) (A) and 17β-estradiol (E2) (B).
Genomic component analysis of S. nematodiphila DH-S01
The genome of this strain consists of a single circularized chromosome. The total length of the genome sequence is 52,56,558 bp, and a total of 4874 encoded genes were annotated. The total length of the gene accounts for 86.14% of the total length of the genome. ANI analysis was performed on the genomic sequences of strain DH-S01 and the genomic sequences of 14 representative Serratia strains uploaded to NCBI (top 10). This strain was shown to be closest to S. nematodiphila DZ0503SBS1 (GenBank ID: NZ_JPUX00000000.1) with an ANI value of 99.55% (). According to the standard proposed by Chun et al. [Citation31], an ANI value above 96% can be used to confirm that it belongs to the same species. Therefore, Serratia sp. DH-S01 was confirmed as S. nematodiphila.
Table 2. Average nucleotide identity analysis (ANI) of Serratia nematodiphila DH-S01.
The genome contains a wide range of functional regions. In addition to the coding region, non-coding regions have the following regulatory functions: transcriptional, post-transcriptional, translational and epigenetic regulation. Several functional regions are also related to the diversity of species evolution. The predicted ncRNAs in S. nematodiphila DH-S01 include 89 tRNAs, 22 rRNAs and 29 sRNAs. Non-coding RNA mainly regulates gene transcription, and the transcription process directly or indirectly affects gene expression and life activities of an organism [Citation19, Citation32]. The concept of the gene island was first proposed by Hacker et al. [Citation33]. Growing evidence suggests that gene islands are involved in the genetic evolution of microorganisms. S. nematodiphila DH-S01 predicted 12 gene islands, each of which can be associated with multiple biological functions such as pathogenic mechanisms and adaptation of organisms.
S. nematodiphila DH-S01 gene function annotation and classification
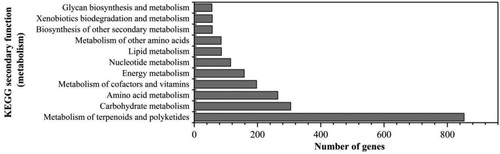
The gene function annotation results of COG, GO, KEGG, NR, Swiss-Prot, TCDB and CAZy were obtained by aligning the coding gene in the S. nematodiphila DH-S01 genome with the database BLAST (Supplemental Table S5). Comparison with the KEGG database showed that a total of 216 metabolic pathways were annotated, and 2233 genes were involved in the metabolic process of this strain (). Among them, a total of 853 genes are involved in the metabolism of terpenoids and polyketides, which account for the highest proportion. Steroids are terpenoid lipids that contain the gonane nucleus of four cycloalkane rings [Citation20]. High abundance of this part of the gene annotation results may be the reason why S. nematodiphila DH-S01 can efficiently degrade steroid estrogen. In addition, carbohydrate metabolism and amino acid metabolism can provide a large amount of energy for the basic growth and metabolic processes of the organism. A total of 305 genes were identified to be involved in carbohydrate metabolism and 264 genes were identified to be involved in amino acid metabolism, both accounting for a higher proportion (). These genes help the bacterial strain to utilize the unique carbon source of steroid estrogen for metabolic processes. S. nematodiphila DH-S01 has annotated 57 genes related to xenobiotics biodegradation and metabolism (), involving 17 types of degradation and metabolism processes, including various aromatic compounds such as benzoate and ethylbenzene.
Figure 3. KEGG metabolic pathway metabolic part classification statistical figure of Serratia nematodiphila DH-S01.
These analyses reflect the xenobiotics degradation ability of strain DH-S01 at the genomic level and indicate S. nematodiphila DH-S01 as a bacterium with abundant metabolic functions and metabolic pathways, which can provide abundant resources for further research on bacterial degradation mechanisms and the development of engineered bacteria with high application value. According to the assembled genomic sequence of the strain, combined with component analysis and functional annotation results of the coding gene, the genome of the strain was visually analysed using Circos software (Supplemental Figure S2).
Analysis of genes encoding steroid estrogen degrading enzymes in S. nematodiphila DH-S01
Based on the annotation results of the KEGG database, a total of 13 predicted coding genes involved in the steroid estrogen degradation process were identified (). Except GM003036, the other 12 genes are annotated to participate in the metabolism of terpenoids and polyketides, involving a total of nine metabolic pathways (map00281, map00903, map01100, map01110, map01120, map01200, map01212, map01130, map01220). This may also indicate a relationship between steroid degradation and the metabolism of terpenoids and polyketides. The size and distribution of these nine oestrogen-degrading enzymes in this strain are shown in . Of these nine steroid estrogen-degrading enzymes identified in this study, seven enzymes belong to steroid side chain oxidases, and one is a steroid core-destroying enzyme.
Figure 4. Location and size of the steroid estrogen-degrading enzyme assumed in Serratia nematodiphila DH-S01.
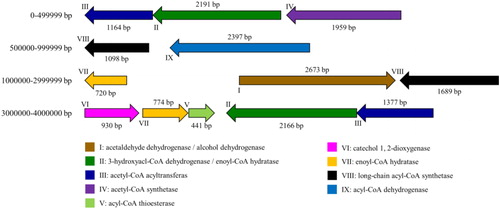
Table 3. Steroid estrogen-degrading enzymes predicted in Serratia nematodiphila DH-S01.
Effective sterol side chain degradation is essential for the production of steroidal intermediates [Citation34]. Steroid side chain degradation is similar to β-oxidation of fatty acids through coenzyme A (CoA) thioester intermediates, generating propionyl and acetyl CoA [Citation37]. The side chain oxidases identified in this study include enoyl-CoA hydratase (echA), acetyl-CoA acyltransferase (fadA), acyl-CoA synthetase (fadD), acyl-CoA dehydrogenase (fadE), etc. Among them, echA, fadD and fadE participate in complete β-oxidation cycle [Citation34], and fadA participates in the degradation of cholesterol side chains [Citation35]. This study also identified acetaldehyde dehydrogenase (hsaG) involved in the A and B loop degradation process [Citation34]. In addition, steroids are substances with a benzene ring, and catechol is an intermediate product of aerobic metabolism of almost all aromatic compounds. The ring opening of catechol is generally achieved by catechol 1,2-dioxygenase and catechol 2,3-dioxygenase [Citation38,Citation39]. In this study, catechol 1,2-dioxygenase (catA) was identified. CatA acts on the carbon bond between the two hydroxyl groups of catechol, which results in ortho-cleavage of catechol and maleic acid is formed [Citation40]. These enzymes form the basis for the efficient degradation of steroid estrogen by strain DH-S01.
Expression of steroid estrogen degrading enzymes in S. nematodiphila DH-S01
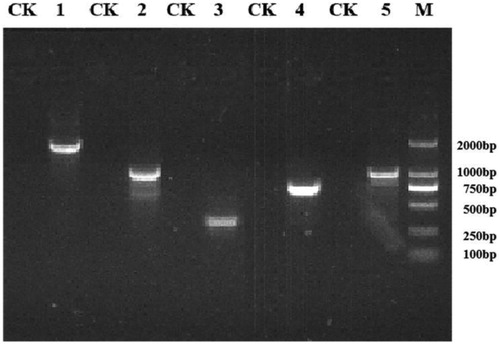
Primers were designed based on the obtained genome-wide sequencing results, and PCR and RT-PCR were performed on the nine steroidal estrogen-degrading enzymes present in S. nematodiphila DH-S01. The PCR amplification results of the corresponding target fragments showed that the bands of these nine enzymes were clearly visible, and the size of each gene fragment was consistent with the sequence length of the sequencing result (). Under the condition that E1 or E2 were provided as the sole carbon source, the gene expression product was obtained by RT-PCR, and the PCR band size was compared. The results of RT-PCR amplification showed that compared with the control group, acetaldehyde dehydrogenase (hsaG), catechol 1,2-dioxygenase (catA) and the four types of steroid side chain oxidases were obtained under the induction of E1 (), including 3-hydroxyacyl-CoA dehydrogenase/enoyl-CoA hydratase (echA), acetyl-CoA acyltransferase (fadA), acyl-CoA thioesterase (paaI) and long-chain acyl-CoA synthetase (fadD). Acetaldehyde dehydrogenase (hsaG) was not expressed under the induction of E2, but catechol 1,2-dioxygenase (catA) and the above four types of steroid side chain oxidases were expressed (). If other steroid estrogens are used, additional expressions of other enzymes may be obtained.
Figure 5. Agarose gel electrophoresis of PCR assay for detection of steroid estrogen degrading enzyme genes.
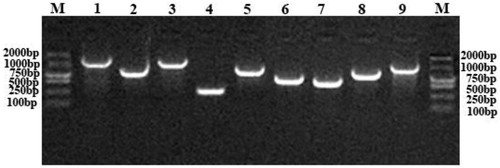
Figure 6. Agarose gel electrophoresis of RT-PCR assay for detection of estrogen degrading enzyme expression in the presence of E1.
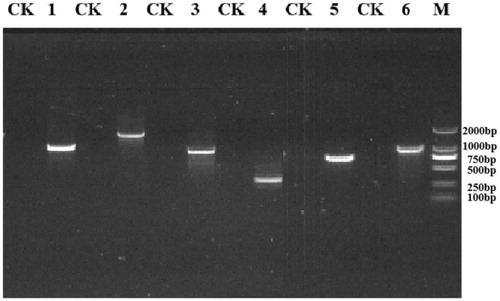
Figure 7. Agarose gel electrophoresis of the RT-PCR assay for detection of estrogen degrading enzyme expression in the presence of E2.
Comparative genomic analysis of S. nematodiphila DH-S01
MAUVE was used to compare and visualize the complete genome sequence of S. nematodiphila DH-S01 and three other strains of Serratia that have been sequenced to confirm the co-linearity of sequences between genomes [Citation30]. This comparison of the four strains showed that S. nematodiphila DH-S01 has higher colinearity with S. nematodiphila DZ0503SBS1 and S. marcescens Db11 and the least linearity with S. fonticola DSM 4576 (). This confirms the afore-mentioned kinship analysis (). In addition, there were duplications of gene fragments and gene level transfer events in these four Serratia strains. Studies have shown that the horizontal transfer of genes may play an important role in the evolution of population structure within bacterial communities and may promote the process of species formation [Citation41]. Duplication of genomic fragments can cause genome size, permutation and function of the loaded genes, as well as changes in interrelationships.
Figure 8. Genomic colinearity analyses (A) of S. nematodiphila DH-S01 and S. nematodiphila DZ0503SBS1, S. fonticola DSM 4576 and S. marcescens Db11. Venn diagram (B) showing the number of clusters of orthologous genes shared and unique between strains clustered in DH-S01, DZ0503SBS1, DSM 4576 and Db11.
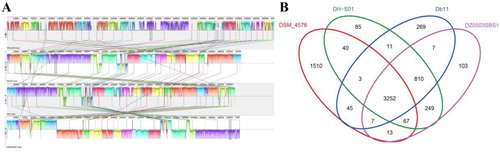
The characteristics of S. nematodiphila DH-S01 and other reference genomes are shown in . Four Serratia strains have similar GC content, coding genes and ncRNA. The Venn diagram () shows 3,252 genes shared by four Serratia strains. S. nematodiphila DH-S01 shares 249, 11 and 40 genes exclusively with S. nematodiphila DZ0503SBS1, S. marcescens Db11 and S. fonticola DSM 4576, respectively. S. nematodiphila DH-S01 has a total of 85 unique genes, which mainly encode hypothetical proteins (Supplemental Table S6). In addition, these unique genes include cold shock protein YdfK-related genes, transcription regulatory protein RcsB-related genes, melamine deaminase-related genes and 2-nitroimidazole transporter-related genes. This part of the gene may be related to the specific environmental adaptability of S. nematodiphila DH-S01, which helps to form the diversity of this species. After searching and statistical analyses (Supplemental Table S7), the genes encoding eight types of steroid estrogen degrading enzyme in belong to the common genes of the four strains. However, catechol 1,2-dioxygenase exists only in S. nematodiphila DH-S01 and S. nematodiphila DZ0503SBS1. It has been speculated that the other three Serratia strains may also have the potential to degrade steroid estrogen.
Table 4. Genomic features of Serratia nematodiphila DH-S01 and other Serratia spp.
Conclusions
Through whole genome sequencing and comparative genomic analysis, we studied the genomic characteristics and genetic features of the steroid estrogen-degrading bacterium. In this study, a strain capable of degrading the steroid estrogen oestrone (E1) and 17β-estradiol (E2) was isolated from a sewage treatment plant. Degradation experiments showed that the degradation rates of E1 and E2 reached 93.47% and 93.2% after 4 days of incubation, respectively. The whole strain was sequenced and identified as S. nematodiphila DH-S01. The genome of the strain consists of a single circular chromosome with a size of 52,56,558 bp, with a total of 4,874 coding genes, and a large number of genes are involved in terpenoid and polyketide metabolism. The related genes of S. nematodiphila DH-S01 degrading steroid estrogen were screened, and the existence and expression of nine types of steroid estrogen degrading enzymes encoded by these genes were verified by PCR and RT-PCR. Comparative genomic analysis showed that S. nematodiphila DH-S01 shared 3,252 genes with three other Serratia strains, including genes encoding eight types of steroid estrogen degrading enzymes. This study confirmed the ability of this strain to degrade steroid estrogen at the genetic level. This not only enriches the genetic information of the genus Serratia, but also provides further information for research on the degradation mechanism of steroid estrogen and degradation-related enzymes.
Supplemental Material
Download MS Excel (20.1 KB)Supplemental Material
Download PDF (390.9 KB)Disclosure statement
The authors report no conflict of interest.
Additional information
Funding
Related Research Data
References
- Adeel M, Song X, Wang Y, et al. Environmental impact of estrogens on human, animal and plant life: A critical review. Environ Int. 2017;99:107–119.
- Robinson BJ, Hellou J. Biodegradation of endocrine disrupting compounds in harbour seawater and sediments. Sci Total Environ. 2009;407(21):5713–5718.
- Thum T, Erpenbeck VJ, Moeller J, et al. Expression of xenobiotic metabolizing enzymes in different lung compartments of smokers and nonsmokers. Environ Health Perspect. 2006;114(11):1655–1661.
- Kidd KA, Blanchfield PJ, Mills KH, et al. Collapse of a fish population after exposure to a synthetic estrogen. Proc Natl Acad Sci USA. 2007;104(21):8897–8901.
- Vajda AM, Barber LB, Gray JL, et al. Reproductive disruption in fish downstream from an estrogenic wastewater effluent. Environ Sci Technol. 2008;42(9):3407–3414.
- Jiang J-Q, Zhou Z, Sharma VK. Sharma Occurrence, transportation, monitoring and treatment of emerging micro-pollutants in waste water — A review from global views. Microchem J. 2013;110:292–300.
- Laurenson JP, Bloom RA, Page S, et al. Ethinyl estradiol and other human pharmaceutical estrogens in the aquatic environment: A review of recent risk assessment data. AAPS J. 2014;16(2):299–310.
- Rajapakse N, Ong D, Kortenkamp A. Defining the impact of weakly estrogenic chemicals on the action of steroidal estrogens. Toxicol Sci. 2001;60(2):296–304.
- Andaluri G, Suri RPS, Kumar K. Occurrence of estrogen hormones in biosolids, animal manure and mushroom compost. Environ Monit Assess. 2012;184(2):1197–1205.
- Biswas S, Shapiro CA, Kranz WL, et al. Current knowledge on the environmental fate, potential impact, and management of growth-promoting steroids used in the US beef cattle industry. J Soil Water Conserv. 2013;68(4):325–336.
- Ray P, Zhao Z, Knowlton KF, KebreabE,. Emerging contaminants in livestock manure: hormones, antibiotics and antibiotic resistance genes. In: Kebreab E, editor. Sustainable animal agriculture. Wallingford: CABI; 2013. p.268–283.
- Combalbert S, Hernandez-Raquet G. Occurrence, fate, and biodegradation of estrogens in sewage and manure. Appl Microbiol Biotechnol. 2010;86(6):1671–1692.
- Wang Y, Shao H, Zhu S, et al. Degradation of 17β-estradiol and products by a mixed culture of Rhodococcus equi DSSKP-R-001 and Comamonas testosteroni QYY20150409. Biotechnol. Biotechnol. Equip. 2019;33(1):268–277.
- Yu CP, Deeb RA, Chu KH. Microbial degradation of steroidal estrogens. Chemosphere. 2013;91(9):1225–1235.
- Roesch LF, Fulthorpe RR, Riva A, et al. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007;1(4):283–290.
- McLellan SL, Huse SM, Mueller-Spitz SR, et al. Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ Microbiol. 2010;12(2):378–392.,
- Zhang T, Shao MF, Ye L. 454 pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 2012;6(6):1137–1147.
- Fujii K, Kikuchi S, Satomi M, et al. Degradation of 17 beta-estradiol by a gram-negative bacterium isolated from activated sludge in a sewage treatment plant in Tokyo, Japan. AEM. 2002;68(4):2057–2060.
- Wu K, Lee TH, Chen YL, et al. Metabolites involved in aerobic degradation of the A and B rings of estrogen. Appl Environ Microbiol. 2019;85:15.
- Donova MV, Egorova OV. Microbial steroid transformations: current state and prospects. Appl Microbiol Biotechnol. 2012;94(6):1423–1447.
- Dovbnya DV, Egorova OV, Donova MV. Microbial side-chain degradation of ergosterol and its 3-substituted derivatives: A new route for obtaining of deltanoids. Steroids. 2010;75(10):653–658.
- Purushotham P, Arun PVPS, Prakash JSS, et al. Chitin Binding Proteins Act Synergistically with Chitinases in Serratia proteamaculans 568. Plos One. 2012;7(5):e36714.
- Weise T, Thürmer A, Brady S, et al. VOC emission of various Serratia species and isolates and genome analysis of Serratia plymuthica 4Rx13. FEMS Microbiol Lett. 2014;352(1):45–53.
- Gutiérrez-Román MI, Holguín-Meléndez F, Dunn MF, et al. Antifungal activity of Serratia marcescens CFFSUR-B2 purified chitinolytic enzymes and prodigiosin against Mycosphaerella fijiensis, causal agent of black Sigatoka in banana (Musa spp.). BioControl. 2015;60(4):565–572.
- Qiu Q, Wang P, Kang H, et al. Genomic analysis of a new estrogen-degrading bacterial strain, Acinetobacter sp. DSSKY-A-001. Int J Genomics. 2019;2019:1–13.
- Li RQ, Zhu HM, Ruan J, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20(2):265–272.
- Li R, Li Y, Kristiansen K, et al. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24(5):713–714.
- Michael R, Ramon RM. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA. 2009;106:19126–19131.
- Wu H, Liu W, Shi L, et al. Comparative genomic and regulatory analyses of natamycin production of Streptomyces lydicus A02. Sci Rep. 2017;7(1):9114.
- Darling AE, Mau B, Perna NT. Progressive Mauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5(6):e11147–4579.
- Chun J, Oren A, Ventosa A, et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol. 2018;68(1):461–466.
- Chen YL, Yu CP, Lee TH, et al. Biochemical mechanisms and catabolic enzymes involved in bacterial estrogen degradation pathways. Cell Chem Biol. 2017;24(6):712–724.e717.
- Lu BX, Leong HW. Computational methods for predicting genomic islands in microbial genomes. Comp Struct Biotechnol J. 2016;14:200–206.
- Van der Geize R, Yam K, Heuser T, et al. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc. Natl Acad Sci USA. 2007;104(6):1947–1952.
- Wilbrink MH, van der Geize R, Dijkhuizen L. Molecular characterization of Itp3 and Itp4, essential for C24-branched chain sterol-side-chain degradation in Rhodococcus rhodochrous DSM 43269. Microbiology-Sgm. 2012;158(12):3054–3062.
- Bragin EY, Shtratnikova VY, Dovbnya DV, et al. Comparative analysis of genes encoding key steroid core oxidation enzymes in fast-growing Mycobacterium spp. Strains. J Steroid Biochem Mol Biol. 2013;138:41–53.
- Crowe AM, Casabon I, Brown KL, et al. Catabolism of the last two steroid rings in Mycobacterium tuberculosis and other bacteria. mBio. 2017;8(2):e00321–17.
- Higson FK, Focht DD. Bacterial degradation of ring-chlorinated acetophenones. Appl Environ Microbiol. 1990;56(12):3678–3685.
- Ghosal D, Dutta A, Chakraborty J, et al. Characterization of the metabolic pathway involved in assimilation of acenaphthene in Acinetobacter sp. strain AGAT-W. Res Microbiol. 2013;164(2):155–163.
- Williams PA, Sayers JR. The evolution of pathways for aromatic hydrocarbon oxidation in Pseudomonas. Biodegradation. 1994;5(3-4):195–217.
- Polz MF, Alm EJ, Hanage WP. Horizontal gene transfer and the evolution of bacterial and archaeal population structure. Trends Genet. 2013;29(3):170–175.