
Abstract
Lung squamous cell carcinoma (LUSC) is highly malignant with poor prognosis, causing prevalent mortality worldwide. Early-stage lung cancer can be distinguished through reliable early detection to urge timely treatment. Non-coding RNAs (ncRNAs) might play such a vital role in tumor initiation, progression and metastasis that they are becoming an important biomarker in cancer screening. To identify the relation between ncRNAs and the early-stage of LUSC, we analyzed the data from the Cancer Genome Atlas (TCGA) database. We identified 28 microRNAs (miRNAs) and 32 long non-coding RNA (lncRNA) that were specifically expressed in early-stage LUSC, and the relationship was shown by the lncRNA-miRNA-mRNA competing endogenous RNA (ceRNA) network. The result derived from Kaplan–Meier survival curves analysis showed that two miRNAs (hsa-miR-22-3p and hsa-miR-552-3p) and a lncRNA (FLJ36000) were negatively correlated with overall survival, but two miRNAs (hsa-miR-27b-5p and hsa-miR-340-5p) and five lncRNAs (CHK8-CPT1B, LINC00167, S NHG15, TTC28-AS1 and WASH7P) were positively associated. Our study provides novel insight for better understanding of early-stage LUSC and facilitates the identification of potential biomarkers for diagnosis and prognosis of early-stage LUSC.
Introduction
Lung cancer, one of the most common cancers all over the world, is becoming the leading cause of cancer-related death and has resulted in 1.8 million deaths worldwide in 2018 [Citation1]. Lung cancer is sub-divided into two main histological types: small cell lung cancer and non-small cell cancer, where the latter contributes for nearly 85% of lung cancer [Citation2]. Non-small cell lung cancer includes lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) [Citation3]. LUSC accounts for approximately 30% of all non-small cell cancer, and about 400,000 deaths annually. It is highly malignant with poor prognosis, and the 5-year survival rate is even less than 15% [Citation4]. Although some oncogenes and suppressor genes have been shown to partly mediate metabolic pathways of LUSC [Citation5], the molecular mechanisms involved are still not clear, particularly the genes that are special or dominant in the regulation of the metabolic network of “born to be bad” primary tumor.
Early-stage lung cancer can be identified through reliable early detection and cured by surgical resection. Thus, it is crucial to describe the specific gene expression profile of early-stage lung cancer to develop prospective biomarkers for early diagnosis and therapeutic targets. Increasing evidence has shown that ncRNAs play key roles in cancer [Citation6]. NcRNAs can be divided into several types, including ribosomal RNA (rRNA), transfer ribonucleic acid (tRNA), microRNAs (miRNAs), long non-coding RNA (lncRNA) and so on [Citation7]. LncRNAs are a collection of more than 200 nucleotide RNAs in length and lack protein-coding function. They are important for many biological processes, including regulating gene expression by regulating chromatin remodeling, controlling post-transcriptional gene expression, processing protein function and locating [Citation8]. A series of notable reports have shown that lncRNAs participate in post-transcriptional gene expression regulation in plants and animals [Citation9, Citation10].
The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/) is the largest public database of tumors worldwide, which provides a variety of histological data for common tumors and becomes an important source for the studies of oncological data currently. We have previously analyzed aberrant gene profiles associated with the progression of LUSC [Citation11]. In this research, we analyzed the relationship among lncRNA, miRNA and mRNA of early-stage LUSC and constructed their competing endogenous RNA (ceRNA) network. Our study focused on specific genes related to the prognosis of LUSC patients, with the characterization of miRNA and lncRNA of early-stage LUSC finally represented.
Materials and methods
RNA sequence data processing and database analysis
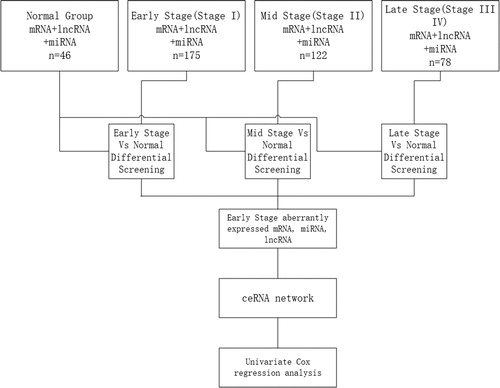
The available data of 375 LUSC tumors and 46 samples of adjacent non-tumorous lung tissues microRNA-seq and RNA-seq were downloaded from TCGA database [Citation11]. The data files at level 3 for a single sample need to be integrated and processed for data analysis. The flow chart for bioinformatics analysis is depicted in . First, the samples were divided into 4 groups according to clinical information, respectively, an early-stage group (Stages I, IA and IB according to histopathological analysis), mid stage group (Stages II, IIA and IIB), late-stage group (Stages III, IIIA, IIIB and IV), and control group with 46 samples of adjacent non-tumorous lung tissues. Second, different expression patterns of mRNAs, miRNAs and lncRNAs were compared among the early-stage group, mid-stage group and late-stage group, respectively, versus the control group. Third, specific series of genes were identified from data of abnormally expressed genes obtained in the early-stage group after common abnormally expressed genes from the 4 groups were filtered.
Figure 1. Flow chart of bioinformatics analysis.
Clinical features of key members of the microRNAs and lncRNAs
After we identified specific abnormally expressed genes only deriving from the early-stage group, we analyzed the clinical features for assessment of the patients. The Cox proportional hazards regression model was employed to analyze the association between these miRNAs, lncRNAs and the LUSC patient survival periods obtained from TCGA. Statistically significant miRNAs and lncRNAs affecting the survival period (p < 0.05) were then determined by Cox regression univariate analysis to construct the Kaplan–Meier survival curve for patients with LUSC subsequently.
Construction of ceRNA network on early-stage LUSC
The lncRNA-miRNA-mRNA ceRNA network was constructed based on the sequestering and binding relationship among differently expressed lncRNA, miRNA and mRNA in the early stage (FDR < 0.05, and p < 0.05). The miRNA seed sequences were determined by mapping the TCGA miRNA identifiers to miRBase (www.miRBase.org, release_21). Using miRanda (http://www.microrna.org/) and Targetscan (http://www.targetscan.org/), and the differently expressed miRNAs that are the mRNA target genes of this study were predicted. The miRanda platform (http://www.microrna.org/) was also applied to predict the lncRNAs targeted by miRNAs. Cytoscape v3.0 was used to construct the network.
Statistical analysis
All data were presented as mean values with standard deviation (± SD) and statistically compared following Student’s t test to compare two groups and analysis of variance (ANOVA) for multiple comparisons. The significance level was set to be 0.05 as default to control the false discovery rate (FDR). Differences were considered statistically significant at p < 0.05.
Results and discussion
Identification of differentially expressedmiRNAs and lncRNAs in early-stage of LUSC
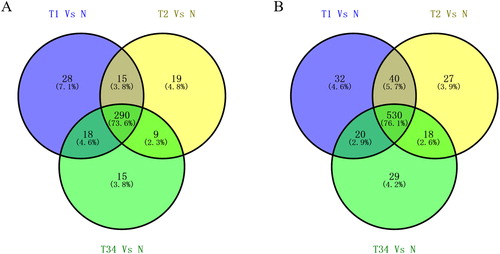
We identified specific expression of 394 miRNAs and 696 lncRNAs from the TCGA database. Analysis of the lncRNA and miRNAs expression profiles in the tissues of LUSC patients and their comparison with adjacent non-tumor lung tissues identified 394 aberrantly expressed miRNAs and 696 lncRNAs. Among these RNAs, we compared the RNAs specially expressed in the early stage with those in other stages of LUSC. Venn diagrams were made to show the relationship among different stages of LUSC (). From the Venn diagrams, 28 miRNAs and 32 lncRNAs were identified which were specially expressed in early stage of LUSC.
Figure 2. Venn diagram analysis of aberrantly expressed lncRNAs (A) and miRNAs (B) between T1 stage/Normal, T2 stage/Normal, T3 stage/Normal and T4 stage/Normal. T1, T2 and T3 represent the early-stage group, the mid-stage group and the late-stage group, respectively.
The feature of aberrantly expressed miRNAs and lncRNAs of early-stage of LUSC
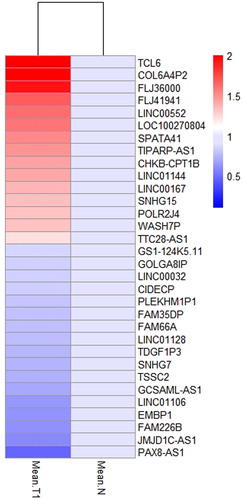
Sixty aberrantly expressed ncRNAs with differential expression levels in 28 miRNAs and 32 lncRNAs are shown in and . Twenty-eight miRNAs contained 9 up-regulated and 19 down-regulated genes, wehreas 32 lncRNAs included 15 up-regulated and 17 down-regulated genes, which represent the characteristics of aberrant expression of microRNAs and lncRNAs in early-stage of LUSC ().
Figure 3. List of 28 differentially expressed miRNAs between normal tissues and T1 stage tissues in LUSC. T1 represents the early stage group. The data are shown as mean values.
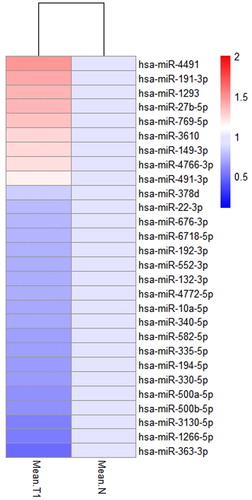
Figure 4. List of 32 differentially expressed lncRNAs between normal tissues and T1 stage tissues in LUSC. T1 represents the early stage group. The data are shown as mean values.
Key miRNAs and lncRNAs and their associated clinical features
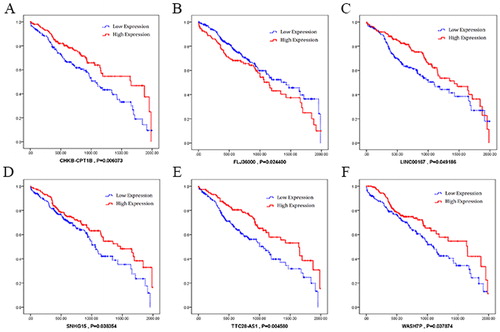
We selected the obtained miRNAs and lncRNAs in early-stage of LUSC to analyse the clinical features associated with their expression level. The Kaplan–Meier survival curves were made to show the results (). The results showed that hsa-miR-22-3p and hsa-miR-552-3p were negatively associated with overall survival; whereas hsa-miR-27b-5p and hsa-miR-340-5p were positively associated (). As for lncRNAs (), CHK8-CPT1B, LINC00167, SNHG15, TTC28-AS1 and WASH7P are positively associated with overall survival, whereas FLJ36000 was negatively associated.
Figure 5. Kaplan–Meier survival curves for four miRNAs (hsa-miR-22-3p, hsa-miR-27b-5p, hsa-miR-340-5p and hsa-miR-552-3p) associated with overall survival. Horizontal axis: overall survival time, days; Vertical axis: survival function.
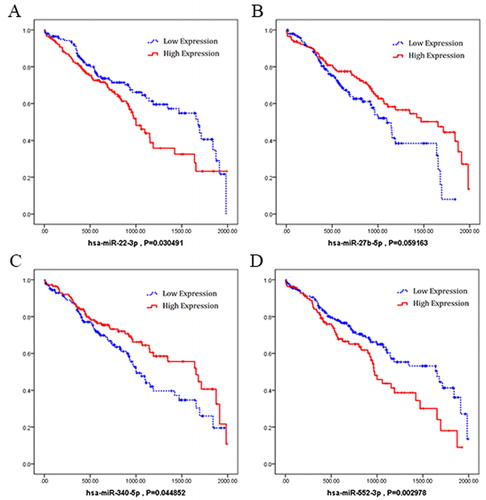
Figure 6. Kaplan–Meier survival curves for six lncRNAs (CHK8-CPT1B, FLJ36000, LINC00167, SNHG15, TTC28-AS1 and WASH7P) associated with overall survival. Horizontal axis: overall survival time, days; Vertical axis: survival function.
Construction of ceRNA network in early-stage of LUSC
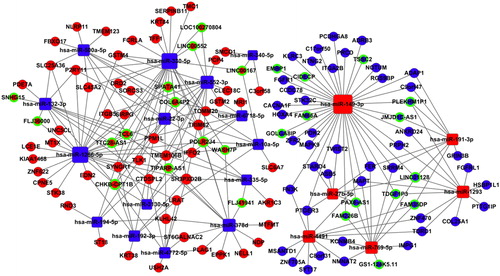
To further understand the pathogenesis of early-stage LUSC, we selected aberrantly expressed 32lncRNAs (17 down-regulated and 15 up-regulated ones) and 28 miRNAs (19 down-regulated and 9 up-regulated ones) in the intersection with 189 mRNA [Citation11] to build an lncRNA-miRNA-mRNA ceRNA network ().
Figure 7. The ceRNA networks in LUSC. Blue squares represent the down-regulated miRNAs; blue balls represent the down-regulated mRNAs; blue balls surrounded by green rings represent the down-regulated lncRNAs. Red diamonds represent the up-regulated miRNAs; red balls represent the up-regulated mRNAs; red balls surrounded by green rings represent the up-regulated lncRNAs.
The obtained results throw more light on the underlying mechanisms implicated in early stage LUSC. Over the past decades, several major mechanisms have been documented, including molecular changes that may contribute to the development and progression of LUSC and LUAD tumors [Citation12], but the average 5-year survival rate of lung cancer is not optimistic yet [Citation13, Citation14]. Thus, in order to improve the survival rate of lung-cancer patients, more and more researchers focus on the identification of cancer-related genes to identify the precise regulatory mechanism of lung cancer initiation and development.
Many cases have shown that a delayed diagnosis will lead to a poorer a prognosis [Citation15]. In recent years, increasing reports have shown that RNAs, which include lncRNAs and miRNAs, are related to human tumors [Citation6]. NcRNAs (including miRNAs and lncRNAs) can be used for early diagnosis in many diseases [Citation16, Citation17]. In this case, we explored the miRNAs and lncRNAs in early stage of LUSC. We analyzed 421 tissues data from TCGA database, including tumor tissues and adjacent non-tumor tissues. By comparing the RNAs expression level among early stage, middle stage, late stage and adjacent tissues, we singled out 28 miRNAs and 32 lncRNAs according to their gene expression level specifically expressed in the early stage of LUSC.
Subsequently, we explored 4 miRNAs and 6 lncRNAs which possess significant relevance to prognosis of LUSC by Kaplan-Meier survival curves analysis. Previous researches showed that hsa-miR-22-3p may be a biomarker in cortisol-producing adrenocortical tumors [Citation18]; hsa-miR-27b-5p might contribute to gastric adenoma [Citation19]; and the up-regulation of mir-340-5p might promote the progression of thyroid cancer [Citation20]. Among the 6 lncRNAs, SNHG15 was reported as a potential prognostic biomarker for hepatocellular carcinoma [Citation21]. Those reports help us to identify the possible roles of a biomarker for early diagnosis of LUSC more easily. In this study, we also found that hsa-miR-552-3p, CHK8-CPT1B, FLJ36000, LINC00167 and TTC28-AS1 were significantly correlated with the prognosis of LUSC, although few reports showed their roles in other cancers. More studies should be carried out to prove their value as potential biomarkers of LUSC in future.
CeRNA is often regarded as a competitive combination component which explains the relationship among lncRNA, miRNA and mRNA. It is generally assumed that lncRNAs and mRNAs can competitively bind miRNAs and can communicate with each other through sharing miRNAs, which represents a new way of gene regulation and plays an important role in the physiological and disease development process [Citation22]. Under certain circumstances, pseudogenes, mRNA, 3′ UTRs, lncRNA and circRNA can all participate in the process of cancer development as ceRNA [Citation23]; the expression level of mRNA will increase or decrease when the miRNA is bonded by lncRNA/circRNA.
Great interest about ceRNA has been aroused in the research of cancer mechanisms, with most of the research in the field verifying how ceRNA expression disorders can affect the progression and pathogenicity [Citation24, Citation25]. Therefore, ceRNA will be a useful tool to elucidate tumor development, and to understand the function of special genes to tumor as a whole [Citation26]. For this purpose, genes that have strong potential regulatory ability will be found by calculating the connectivity of lncRNA/mRNA within the network. For example, Yin et al.’s research revealed that tumor progression was promoted by the lncRNA SNHG6-003, which functions as a ceRNA in hepatocellular cancer [Citation27]. Le et al.’s results suggest that tumor specific mRNAs and lncRNAs may be involved in complex ceRNA networks, which are potential targets for triple-negative breast cancer (TNBC) therapy [Citation28]. Besides, the results reported by Hyun et al. [Citation29] suggested that 3′ UTR shortening plays a major role in the inhibition of tumor suppressor genes in trans cells by interfering with ceRNA crosstalk. These studies showed that the research of ceRNA is an essential part of cancer research.
In this study, hundreds of candidates from TCGA database were used to construct a ceRNA network to reveal a novel regulatory network during the early stage of LUSC, with the relationship between LUSC and the mRNA, miRNA, lncRNA identified as a result. Our findings may provide a novel research direction for better understanding of miRNA and lncRNA-related ceRNA networks of LUSC, as well as propose a valuable insight into the development of the early-stage of LUSC.
Conclusions
Screening on the early-stage LUSC provides a favorable prognosis for LUSC patients. We identified specific miRNAs and lncRNAs only significantly expressed in the early-stage of LUSC, among which 4 specific miRNAs and 6 lncRNAs were significantly correlated to the clinical survival rate. Our finding may reveal a potential biomarker for screening LUSC. Meanwhile, we have constructed a ceRNA network of the early-stage LUSC, which may contribute to better understanding of the related pathological mechanism.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Freddie B, Jacques F, Isabelle S, et al. Global cancer statistics 2018: globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clinicians. 2018;68(6):394–424.
- Song X, Kong F, Zong Z, et al. Mir-124 and mir-142 enhance cisplatin sensitivity of non-small cell lung cancer cells through repressing autophagy via directly targeting sirt1. RSC Adv. 2019;9(9):5234–5243.
- Tang RX, Chen WJ, He RQ, et al. Identification of a RNA-seq based prognostic signature with five lncRNAs for lung squamous cell carcinoma. Oncotarget. 2017;8(31):50761–50773.
- Sui J, Xu SY, Han J, et al. Integrated analysis of competing endogenous RNA network revealing lncRNAs as potential prognostic biomarkers in human lung squamous cell carcinoma. Oncotarget. 2017;8(39):65997–66018.
- Bonelli MA, Cavazzoni A, Saccani F, et al. Inhibition of pi3k pathway reduces invasiveness and epithelial-to-mesenchymal transition in squamous lung cancer cell lines harboring pik3ca gene alterations. Mol Cancer Ther. 2015;14(8):1916–1927.
- Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2018;18(1):5–18.
- Sui J, Li YH, Zhang YQ, et al. Integrated analysis of long non-coding RNA-associated ceRNA network reveals potential lncRNA biomarkers in human lung adenocarcinoma. Int J Oncol. 2016;49(5):2023–2036.
- Yi J, Li S, Wang C, et al. Potential applications of polyphenols on main ncRNAs regulations as novel therapeutic strategy for cancer. Biomed Pharmacother. 2019;113:108703. [cited 2020 Jun 03]
- Wang P, Xu J, Wang Y, et al. An interferon-independent lncRNA promotes viral replication by modulating cellular metabolism. Science. 2017;358(6366):1051–1055.
- Xu W, Yang T, Wang B, et al. Differential expression networks and inheritance patterns of long non-coding RNAs in castor bean seeds. Plant J. 2018;95(2):324–340.
- Ning P, Wu Z, Hu A, et al. Integrated genomic analyses of lung squamous cell carcinoma for identification of a possible competitive endogenous RNA network by means of TCGA datasets. Peerj. 2018;6(16):e4254. [cited 2020 Jun 03]
- Wang G, Chen H, Liu J. The long noncoding RNA LINC01207 promotes proliferation of lung adenocarcinoma. American Journal of Cancer Research. 2015;5(10):3162–3173.
- Zhao Y, Lin H, Jiang J, et al. TBL1XR1 as a potential therapeutic target that promotes epithelial‐mesenchymal transition in lung squamous cell carcinoma. Exp Ther Med. 2018;17(1):91–98.
- Wei Y, Zhang X. Transcriptome analysis of distinct long non-coding RNA transcriptional fingerprints in lung adenocarcinoma and squamous cell carcinoma. Tumor Biol. 2016;37(12):16275–16285.
- Petty TL. Early diagnosis of lung cancer. Hosp Pract (1995). 2001;36(5):9–10.
- Zhang Y, Sui J, Shen X, et al. Differential expression profiles of micrornas as potential biomarkers for the early diagnosis of lung cancer. Oncol Rep. 2017;37(6):3543–3553.
- Zheng HT, Shi DB, Wang YW, et al. High expression of lncRNA MALAT1 suggests a biomarker of poor prognosis in colorectal cancer. Int J Clin Exp Path. 2014;7(6):3174–3181.
- Perge P, Decmann Á, Pezzani R, et al. Analysis of circulating extracellular vesicle-associated micrornas in cortisol-producing adrenocortical tumors. Endocrine. 2018;59(2):280–288.
- Kim YJ, Hwang KC, Kim SW, et al. Potential mirna-target interactions for the screening of gastric carcinoma development in gastric adenoma/dysplasia. Int J Med Sci. 2018;15(6):610–616.
- Zhao P, Ma W, Hu Z, et al. Up-regulation of mir-340-5p promotes progression of thyroid cancer by inhibiting bmp4. J Endocrinol Invest. 2018;41(10):1165–1172.
- Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21(6):354–361.
- Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. 2016;17(5):272–283.
- Karreth FA, Pandolfi PP. ceRNA cross-talk in cancer: when ce-bling rivalries go awry. Cancer Discov. 2013;3(10):1113–1121.
- Karreth FA, Reschke M, Ruocco A, et al. The BRAF pseudogene functions as a competitive endogenous RNA and induces lymphoma in vivo. Cell. 2015;161(2):319–332.
- Poliseno L, Pandolfi PP. PTEN ceRNA networks in human cancer. Methods. 2015;77–78:41–50.
- Cian G, Aoife ML. Pseudogenes provide evolutionary evidence for the competitive endogenous RNA hypothesis. Mol Biol Evol. 2018;35(12):2886–2899.
- Yin H, Wang X, Zhang X, et al. Integrated analysis of long noncoding RNA associated-competing endogenous RNA as prognostic biomarkers in clear cell renal carcinoma. Cancer Sci. 2018;109(10):3336–3349.
- Kehao L, Hui G, Qiulei Z, et al. Gene and lncRNA co-expression network analysis reveals novel ceRNA network for triple-negative breast cancer. Sci Rep. 2019;9(1):15122. [cited Jun 03]
- Hyun JP, Ping J, Soyeon K, et al. 3′ UTR shortening represses tumor-suppressor genes in trans by disrupting ceRNA crosstalk. Nat Genet. 2018;50(6):783–789.