
Abstract
This report examines the soluble expression of a novel feruloyl esterase, BpFae, from Burkholderia pyrrocinia in Escherichia coli overexpression systems using three expression vectors, pET28a, pCold-TF and pGEX-4T-1. BpFae was overexpressed as insoluble inclusion bodies when pET28a was used as the expression vector. BpFae was overexpressed in the soluble form when using pCold-TF; however, the overexpressed protein showed negligible activity. Recombinant BpFae was produced in the soluble form and active when E. coli cells were transformed with the pGEX-4T-1-BpFae vector. Optimal conditions for soluble overexpression of recombinant BpFae were determined, and the highest activity of recombinant BpFae was 2.54 U mL−1. Multiple sequence alignment, phylogenetic analysis and construction of a three-dimensional model indicated that BpFae is a unique FAE from B. pyrrocinia with underlying research value.
Introduction
Feruloyl esterases (FAEs, EC 3.1.1.73) constitute a subclass of carboxylic acid esterases that release phenolic acids, such as ferulic acid (FA) and their dimers, from lignocellulosic plant biomass by hydrolyzing ester bonds between hydroxycinnamic acids and plant polysaccharides [Citation1,Citation2]. With the ability to remove hydroxycinnamic acids from plant cell walls and synthesize ester-linked hydroxycinnamic acids, FAEs have potentially important roles in biotechnological processes for various industrial applications [Citation3]. In the pulp and paper industry, FAEs are used with bleaching pulp to lower energy consumption [Citation4]. In the feed industry, FAEs improve access of main chain degrading enzymes to facilitate fibre digestion and bioavailability of phytonutrients [Citation3]. In addition, FAEs are essential accessory enzymes to complete hydrolysis of lignocellulosic biomass for bioethanol and other biorefineries. FAEs have been used in the food industry to remove both off-flavors/odors and to enhance the aroma of several seasonings and alcoholic beverages [Citation3]. FAEs are also suggested to improve dough rheology when added as a supplement in the baking process [Citation1]. The most important applications of FAEs involve degradation of biomass to release FA. FA is a valuable chemical to the cosmetic industry because of its claimed antioxidant, UV-absorbing activity, de-pigmenting activity and role as a carrier of vitamin C and E [Citation5]. Furthermore, FA displays pharmaceutical and health beneficial functions, e.g. antimicrobial, anti-inflammatory, anti-diabetic, anti-thrombosis, anti-cancer and cholesterol-lowering activity [Citation6].
Considering their great potential in industrial and agricultural applications, most FAEs have been identified in plants and microorganisms [Citation7]. Currently, FAEs have been isolated primarily from fungi and only a few studies have examined FAEs from bacteria [Citation8,Citation9]. Cloned FAEs from bacteria are even more scarce and limited to rumen microorganisms, i.e. Butyrivibrio proteoclasticus [Citation10], Cellulosilyticum ruminicola [Citation11] and Lactobacillus (L. amylovorus, L. acidophilus, L. farciminis, L. fermentum [Citation8], L. johnsonii [Citation12]), and those of other bacterial origin, including Streptomyces werraensis [Citation1], Streptomyces sp. [Citation13], Actinomyces sp. [Citation14], Clostridium thermocellum [Citation15], Geobacillus thermoglucosidasius [Citation16] and Dickeya dadantii [Citation17]. Bacterial FAEs were easier to express heterologously and more convenient to apply. So, more FAEs of bacterial origin need to be mined. Furthermore, bacterial and fungal FAEs are encoded differently. According to Crepin’s classification based on substrate utilization and amino acid sequence similarities [Citation18], many bacterial FAEs do not belong to any type from the previous four classifications (A, B, C and D), indicating that the functional classification of microbial FAEs based on fungal origin is inappropriate. In previous studies, we screened Burkholderia pyrrocinia (B. pyrrocinia) which showed high lipase activity [Citation19], making it the first report of a lipase from B. pyrrocinia. In addition, the strain efficiently degraded phthalate esters (PAEs) by using a novel biodegradation pathway [Citation20], which is conducted primarily by esterases of B. pyrrocinia. Thus, B. pyrrocinia may be suitable for use in many applications. Therefore, we sequenced the whole genome of B. pyrrocinia. A gene encoding a FAE was identified, and this was the first report of a FAE cloned and characterized from Burkholderia.
Currently, FAE activity of wild-type strains is relatively low [Citation21,Citation22], and production of FAEs by wild-type strains is time consuming [Citation23]. Heterologous expression of target proteins is essential for characterization. Thus, to improve activity of and characterize FAEs it is necessary to heterologously express these proteins. In general, Escherichia coli is the most commonly used expression platform of heterologous proteins. As a well-established cell factory, it grows fast and achieves high cell density cultures easily. Furthermore, the media it needs are inexpensive and easy to get. Transformation with exogenous DNA is fast and easy [Citation24]. Nonetheless, there are a number of issues with using E. coli as the expression host, such as no or low expression, protein inactivity and inclusion body (IB) formation, which results from an unbalanced equilibrium between protein aggregation and solubilization. These problems are overcome by optimization of culture and induction conditions, expression in the cytoplasm, the use of different vectors or engineered hosts, fusion with various functional tags and co-expression of chaperones. A number of expression vector libraries with different features have been developed, e.g. pET, pCold and pGEX. pET vectors with the T7 promoter system are extremely common for recombinant protein expression and remain the most used option for obtaining large amounts of target protein [Citation24]. The pCold-TF plasmid is a fusion cold-shock expression vector that expresses the trigger factor (TF) chaperone as a soluble fusion tag. TF is a 48-kDa prokaryotic ribosome-associated chaperone protein from E. coli that is expressed highly in E. coli expression systems. It can facilitate protein folding correctly, and enable efficient soluble protein at a lowered incubation temperature [Citation25]. Kim et al. [Citation26] produced large quantities of recombinant amyloid-β peptide 1–42 in a soluble form using the pCold-TF vector, and Saini et al. [Citation25] successfully used this vector system to transform insoluble aggregates of recombinant epoxide hydrolases to the soluble and functionally active form. The pGEX-4T-1 vector expresses target proteins with a glutathione S-transferase (GST) tag. The GST tag is best suited for prokaryotic expression because GSTs are a family of multifunctional cytosolic proteins that are present in eukaryotic organisms but generally not found in bacteria [Citation27]. The GST tag is an established affinity tag based on the strong affinity of GST for immobilized glutathione [Citation28], and has long been used to increase the solubility of fusion proteins in E. coli. Yu et al. [Citation29] achieved high expression of soluble FHL2 (four and a half LIM domains 2) as a GST fusion protein in E. coli BL21 cells. Using the pGEX-4T-1 plasmid, Rabhi-Essafi et al. [Citation30] achieved 70% expression of soluble recombinant human interferon α as a GST fusion protein.
In this report, to explore the characteristics and application of the FAE from B. pyrrocinia, E. coli was used as the expression host and standard expression vectors were tested for production of soluble recombinant BpFae. The culture and induction conditions of E. coli-producing BpFae were optimized.
Materials and methods
Materials and strains
B. pyrrocinia B1213 was used as the source of FAE. This bacterium strain was isolated from soil, identified and deposited in the CGMCC (China General Microbiological Culture Collection Center) with the accession number CGMCC, No. 12806 [Citation19]. E. coli strains DH5α and BL21(DE3) (Takara, Shiga, Japan) were used for plasmid preparation and protein expression, respectively. Plasmids pET28a, pCold-TF, pGEX-4T-1 (Takara) were used as the expression vectors. The EZgene Plasmid Miniprep kit and the E.Z.N.A. Gel Extraction Kit were purchased from Biomiga Inc. (San Diego, CA, USA) and Omega Bio-tek, Inc. (Atlanta, GA, USA), respectively. Restriction enzymes and other reagents for gene manipulation were purchased from Takara Bio, Inc.
Cloning and sequencing of the BpFae gene
The whole genome of B. pyrrocinia B1213 was sequenced previously. The gene encoding the BpFae protein was used to design a primer set F/R () for PCR cloning of the BpFae open reading frame (ORF) from B. pyrrocinia B1213. Extraction of genomic DNA from B. pyrrocinia B1213 was carried out using the E.Z.N.A. Bacterial DNA Kit in accordance with the manufacturer’s instructions. LA-Taq polymerase (Takara) was used for amplifying the FAE gene from B. pyrrocinia B1213 genomic DNA. The amplification mix contained 5 µL LA-Taq buffer, 5 µL 2.5 mmol L−1 dNTP mix, 1 µL genomic DNA, 1 µL forward primer (10 mmol L−1), 1 µL reverse primer (10 mmol L−1), 0.5 µL LA-Taq polymerase (2.5 U µL−1) and water to 50 µL. The reaction was performed in a T100 Thermo Cycler (Bio-Rad, Hercules, CA, USA) with 5 min at 94 °C, 30 cycles of 30 s at 94 °C, 1.5 min at 65 °C, 1 min at 72 °C and a final elongation step of 10 min at 72 °C. The PCR product was ligated into the pMD-18T vector (Takara) and sequenced by SinoGenoMax, Beijing, China.
Table 1. Primers used in this study.
Construction of the recombinant plasmids
Using the pMD-18T plasmid containing the BpFae gene as the template, the BpFae gene (1722 bp) with different homologous sequences was amplified by PCR with primers A1/A2, D1/D2 and X1/X2 (), and the PCR products were cloned into the expression vectors pET-28a, pCold-TF and pGEX-4T-1, respectively, using the NovoRec plus One step PCR Cloning Kit (Novoprotein, Shanghai, China).
Bioinformatics analysis and three-dimensional modelling of BpFae
The molecular weight (MW) and isoelectric point (pI) of BpFae were analyzed using the EXPASY site (http://www.expasy.ch). Signal analysis was carried out using the SignalP 4.0 Server. Multiple sequence alignments were performed and displayed using Molecular Evolutionary Genetics Analysis version X (MEGA X) [Citation31] and the programme ESPript 3 (http://espript.ibcp.fr) [Citation32], respectively. Phylogenetic analyses were conducted using MEGA version X [Citation31] with the neighbor-joining method. A three-dimensional (3-D) model of the BpFae structure was performed using Discovery Studio 2.6 (DS 2.6) software (Accelrys, BioVia, San Diego, CA, USA) based on the crystal structure of an MHETase from Ideonella sakaiensis (I. sakaiensis) (PDB ID: 6QG9) [Citation33]. The modelled 3-D structure of BpFae was visualized using PyMOL (http://pymol.org).
Strain cultivation
For expression of BpFae, transformed E. coli BL21 cells were cultured in Luria–Bertani (LB) broth (100 mL) containing 100 μg mL−1 ampicillin (pCold-TF-BpFae and pGEX-4T-1-BpFae) or 40 μg mL−1 of kanamycin (pET28a-BpFae) to an optical density of 0.6–0.8 at 37 °C. The cells were induced with a final concentration of 0.1 mmol L−1 isopropyl-d-thiogalactopyranoside (IPTG) and were grown for an additional 20 h at 20 °C (pGEX-4T-1-BpFae and pET28a-BpFae) and 15 °C (pCold-TF-BpFae).
Extraction of BpFae from cell fractionations
The culture broth was centrifuged at 9400 g for 10 min at 4 °C to separate the supernatant from the cells. The harvested cells were resuspended using lysis buffer (50 mmol L−1 potassium phosphate, pH 7.0). The lysed cells were disrupted by sonication (100 W, 20 kHz pulses: 2 s on, 3 s off; total time: 15 min). The disrupted cells were centrifuged at 15,000 g for 15 min. The supernatant constituted the soluble fraction and the pellet resuspended in lysis buffer constituted the insoluble fraction.
FAE activity assay
Methyl ferulic acid (MFA, 25 mmol L−1, dissolved in DMSO) as the substrate was diluted with 50 mmol L−1 sodium phosphate buffer (pH 7.0) to 1 mmol L−1. The 450 µL substrate solution was pre-incubated at 37 °C for 5 min and then mixed with 50 µL diluted enzyme solution and incubated at 37 °C for 10 min. The reaction was terminated by adding 500 µL acetonitrile [Citation1,Citation34]. The substrates and products were analyzed by high performance liquid chromatography (HPLC) with a UV detector (Agilent, Santa Clara, CA, USA). Samples were filtered through a 0.22-µm filter and applied to a chromatography system equipped with a ZORBAX Eclipse Plus C-18 column (Agilent). The mobile phase was composed of solvent A (acetonitrile) and solvent B (water and acetic acid, 99:1, v/v) with a ratio of 7:3 (v/v), and eluted at a flow rate of 0.6 mL min−1. The column eluent was monitored at 320 nm [Citation34,Citation35].
One unit (U) of enzyme activity was defined as the required enzyme amount for releasing 1 µmol ferulic acid in 1 min under the standard conditions provided above.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
SDS-PAGE was performed under denaturing conditions on a 10% separating gel and 4.5% concentrating gel. Gels were visualized with 0.25% Coomassie Brilliant Blue R-250 staining [Citation36].
Removal of the trigger factor
Soluble fractions of the TF-BpFae fusion protein were diluted to 0.1 mg mL−1 with Tris-HCl buffer (Tris 20 mmol L−1, 150 mmol L−1 NaCl, pH 8.0) and 1 mL of TF-BpFae was cleaved by 0.5 µL (1000 U mL−1) thrombin (Solarbio, Beijing, China) at 20 °C for 5 h.
Zymographic analysis
Zymograms were performed based on a previous report [Citation37]. Native PAGE was carried out using a 7.5% polyacrylamide gel. The soluble protein sample was diluted two-fold by using Tris-glycine buffer (25 mmol L−1 Tris, 200 mmol L−1 glycine, pH 8.3, containing 10% glycerol). Activity was detected by adding 5 mmol L−1 MFA and incubation at 37 °C for 20 min. A yellow band representing feruloyl esterase was observed in the gel.
Optimization of FAE production
Maximum BpFae expression was optimized by testing the following parameters: culture and induction conditions, such as culture media (LB, Terrific Broth medium (TB), Medium for eXpression (MX), buffered Luria-Bertani medium with magnesium (LBBM), buffered Luria-Bertani medium with magnesium and extra NaCl (LBBNM), buffered Luria-Bertani medium with glycerol and extra NaCl (LBBNG), buffered Luria-Bertani medium with magnesium, glycerol and sorbitol (LBBSMG) [Citation38], Terrific Broth medium with glycerol and NaCl (TB-GN) [Citation39] and super optimal broth (SOB) [Citation40]), initial pH values, IPTG concentration, inoculum size, induction initiation time, induction temperature, rotation speed and post-induction time ().
Table 2. Factors and levels for optimization of BpFae production.
Statistical analysis
Each treatment was performed in triplicate and the results are expressed as mean values with standard deviation (±SD). All statistical analyses were performed with OriginPro 8.6 (OriginLab, Northampton, MA, USA) and Excel 2016 (Microsoft, Redmond, WA, USA).
Nucleotide sequence accession number
The nucleotide sequence data for BpFae was submitted to GenBank under the accession number MN848242.
Results and discussion
Cloning of the BpFae gene
A 1722-bp fragment encoding the BpFae from B. pyrrocinia B1213 was isolated via PCR and cloned into pMD18-T successfully. The BpFae gene was then amplified using A1/A2, D1/D2 and X1/X2 primers and successfully cloned into the expression vectors pET-28a, pCold-TF and pGEX-4T-1, respectively. These expression plasmids were transformed into E. coli BL21 (DE3) cells.
Bioinformatics analysis of BpFae
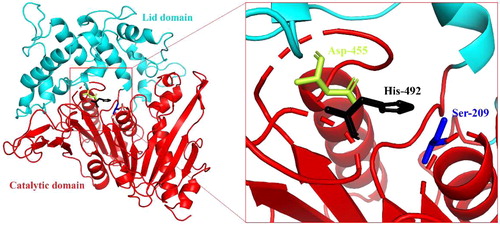
The analysis of the whole gene of B. pyrrocinia B1213 revealed the presence of a 1719-bp open reading frame (ORF) encoding a hypothetical 573 amino acids FAE nominated as BpFae with a MW of 59.04 kDa and a pI of 5.09. SignalP 4.1 server analysis suggested that a signal peptide of 19 amino acids existed. The MW of BpFae is larger than most FAEs from prokaryotes (27–45 kDa) [Citation8,Citation10,Citation12,Citation16,Citation17,Citation41]. In addition, multiple protein sequence alignment between BpFae and three esterases for which crystal structures are available and share the highest sequence identity with BpFae showed that a ‘Gly-x-Ser-x-Gly’ motif was present, which is highly conserved in reported FAEs [Citation16]. The conserved motif for BpFae was G-C-S-N-G (; Gly 207 to Gly 211). FAEs display a common fundamental catalytic triad mechanism consisting of a serine (Ser), a histidine (His) and a carboxylic acid, namely aspartic acid (Asp) or glutamic acid [Citation7]. BpFae has a classical Ser-Asp-His catalytic triad (Ser 209, His 492, Asp 455).
Figure 1. Multiple sequence alignment of BpFae and three similar esterases with known crystal structures on the amino acid level. The aligned sequences were derived by the MEGA X software and visualized by the ESPript 3 programme. MHETase-6QG9 from Ideonella sakaiensis; AoFae-6G21from Aspergillus oryzae; FaeB-3WMT from Aspergillus oryzae. The conserved G-X-S-X-G motif and the putative catalytic triad (S209, D492 and H455) is boxed in and indicated by stars.

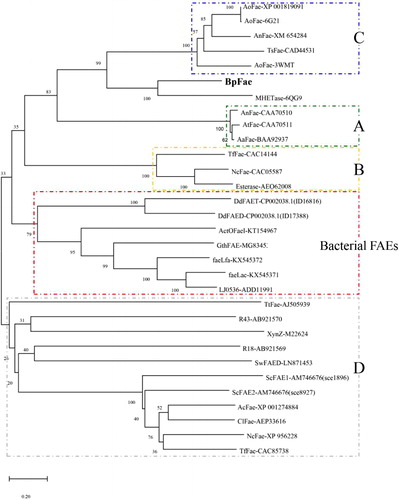
The first phylogenetic tree () depicts the well characterized FAEs derived from bacteria, including SwFAED from Streptomyces werraensis [Citation1], faeLam from L. amylovorus, faeLac from L. acidophilus, faeLfa from L. farciminis, faeLfe from L. fermentum [Citation8], Est1E from Butyrivibrio proteoclasticus [Citation10], CrFAE1, CrFAE2 and CrFAE3 from Cellulosilyticum ruminicola [Citation11], LJ0536 and LJ1228 from L. johnsonii [Citation12], R18 and R43 from Streptomyces sp. [Citation13], ActOFaeI from Actinomyces sp. [Citation14], XynZ from Clostridium thermocellum [Citation15], GthFAE from Geobacillus thermoglucosidasius [Citation16], DdFAED and DdFAET from Dickeya dadantii [Citation17] and ScFAE1 and ScFAE2 from Sorangium cellulosum [Citation41]. Pairwise alignment of FAEs showed no greater identity than 20%, and these bacterial FAEs could be divided into several classes as shown in . BpFae, ScFAE1 and ScFAE2 were grouped into the same cluster, and BpFae was found to have 14% and 12% amino acid identity (DNAMAN 8) with ScFAE1 and ScFAE2 from S. cellulosum, respectively. The relatively low identity to other FAEs underlines the uniqueness of BpFae.
Figure 2. Phylogenetic relationship of BpFae and other well characterized feruloyl esterases derived from bacteria. The tree was constructed using MEGA X with the neighbor-joining algorithm.

The second phylogenetic tree () depicts known FAEs derived from eukaryotic microorganisms including types A, B, C and D, and includes AoFae-XP 001819091 from Aspergillus oryzae [Citation42], AoFae-6G21 from A. oryzae, AnFae-XM654284 from Aspergillus nidulans [Citation43], TsFae-CAD44531.1 from Talaromyces stipitatus [Citation44], AoFae-3WMT from A. oryzae [Citation45], MHETase-6QG9 from I. sakaiensis [Citation33], AnFae-CAA70510 from Aspergillus niger [Citation46], AtFae-CAA70511 from Aspergillus tubingensis [Citation46], AaFae-BAA92937 from Aspergillus awamori [Citation47], TfFae-CAC14144 from Talaromyces funiculosus (Penicillium funiculosum) [Citation48], NcFae-CAC05587 from Neurospora crassa [Citation49], TtFae-AEO62008.1 from Thermothelomyces thermophilus [Citation50], AcFae-XP 001274884 from Aspergillus clavatus [Citation51], ClFae-AEP33616.1 from Chrysosporium lucknowense [Citation52], NcFae-XP 956228 from N. crassa [Citation53], TfFae-CAC85738 from T. funiculosus (P. funiculosum), and some bacterial FAEs present in the first phylogenetic tree (). As shown in , most eukaryotic FAEs were classified only into A, B, C and D types, which is consistent with their individual characteristics described previously [Citation3]. In contrast, most bacterial FAEs were classified into a special type without any FAEs from fungi, suggesting that fungal and bacterial FAEs are encoded differently [Citation7]. Interestingly, BpFae and MHTEase from I. sakaiensis were grouped into the same cluster, which is similar to the type C FAEs and differs greatly from most bacterial FAEs.
Figure 3. Evolutionary relationship of BpFae to feruloyl esterases derived from eukaryotic microorganism including type A, B, C and D, and some bacterial FAEs contained in the first phylogenetic tree (). The tree was constructed using MEGA X with the neighbor-joining algorithm.
Based on the known crystal structure of MHETase from I. sakaiensis (PDB ID: 6QG9), which shares 45.1% sequence identity with BpFae, a 3-D structural model of BpFae was generated by homology modelling (). BpFae consists of two domains: a catalytic domain (residues 38–237 and 432–573) containing the Ser209-His455-Asp492 catalytic triad and a lid domain (residues 238–431) that covers the active site. The catalytic domain adopts a typical α/β-hydrolase fold, and the lid domain inserts between β-strand 7 and α-helix 15 of the α/β-hydrolase fold.
Figure 4. The predicted three-dimensional structure of BpFae by DS sever based on the crystal structure of a MHETase from Ideonella sakaiensis (PDB code 6QG9). The catalytic domain (cyan) and the lid domain (red) are shown. BpFae had a typical α/β hydrolase fold and a Ser-Asp-His catalytic triad (S209, D492 and H455).
Expression of pET28a-BpFae
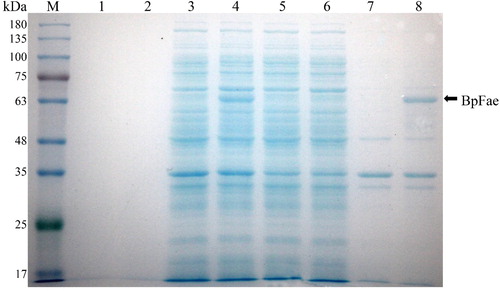
Recombinant BpFae production using the pET28a-BpFae expression plasmid in E. coli BL21 (DE3) cells was confirmed by SDS-PAGE analysis and the observation of a protein band at ∼60 kDa (). This protein band was present in both the total protein and pellet fraction samples, indicating that recombinant BpFae was expressed in an insoluble aggregate or IBs. The FAE activity of the cell fractions was examined. The total protein fraction (0.005 U mL−1) and supernatant (0.005 U mL−1) displayed negligible activity.
Figure 5. SDS-PAGE analysis of the expression of BpFae in E. coli BL21(DE3) using pET28a vector. M: Molecular weight marker. Lanes 1, 3, 5 and 7: BL21(DE3)-pET-28a; lanes 2, 4, 6 and 8: BL21(DE3)-pET-28a-BpFae. Lanes 1 and 2: supernatant of culture medium; lanes 3 and 4: lysate; lanes 5 and 6: supernatant of lysate; lanes 7 and 8: pellet of lysate. The molecular weight of His-BpFae was predicted as 59 kDa, which was consistent with that as seen in the gel.
Recombinant proteins expressed in the cytoplasm of E. coli are often misfolded or aggregated [Citation24] because the newly synthesized target protein is expressed in the microenvironment of E. coli, which may differ from the original source in terms of pH, osmolarity, redox potential, cofactors and folding mechanisms. All of these factors lead to protein instability and aggregation, i.e. IBs. The pET-28a expression vector uses the T7 promoter system, potentially allowing for high levels of expression at high rates [Citation54]. IB formation results from an unbalanced equilibrium between protein aggregation and solubility [Citation24], which often occurs as a stress response when recombinant proteins are expressed at high rates. Therefore, the pET-28a expression vector facilitated formation of inclusion bodies in the process of heterologous protein expression [Citation51]. The most convenient way to ameliorate factors leading to IB formation is to reduce the rate of protein expression, which enables newly transcribed recombinant proteins to fold correctly [Citation24]. Therefore, studies have shown that lowering the induced temperature and reducing the concentration of chemical inducer can shift protein expression to the soluble form [Citation55,Citation56]. Unfortunately, changing either of these parameters did not lead to soluble BpFae expression using the pET28a-BpFae expression plasmid (data not shown).
Expression of pCold-TF-BpFae
As a functional class of unrelated proteins, molecular chaperones can aid protein refolding of stable protein aggregates, assist folding of the newly synthesized polypeptides and allow the evolution of new protein functions and phenotypic traits [Citation57–60]. Co-expression of target proteins with molecular chaperones has been shown to improve production of soluble, correctly folded target proteins [Citation61–63].
In 2004, the pCold expression vector series was developed, which is highly complementary to the widely used pET vectors. These expression vectors are cold-shock vectors where protein expression is under the control of the cspA promoter [Citation64,Citation65]. The pCold-TF DNA vector is a fusion cold-shock expression vector that expresses the Trigger Factor (TF) chaperone as a soluble tag, facilitating co-translational folding of nascent polypeptides and reduction in protein misfolding [Citation66]. Trigger Factor is a prokaryotic ribosome-associated chaperone protein (48 kDa) that is highly expressed in E. coli BL21 (DE3) cells. Furthermore, the vector enables efficient soluble protein expression at lower incubation temperatures, which facilitates effective protein folding and reduces the activity of cellular proteases.
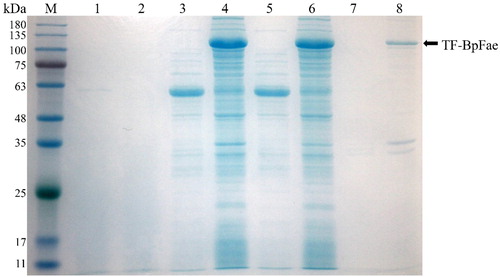
Hence, the pCold-TF-BpFae expression vector was constructed, and the target fusion protein TF-BpFae was overexpressed at 15 °C in E. coli BL21 (DE3) cells. TF-BpFae has a predicted MW of 111 kDa (). The results of SDS-PAGE analysis from total protein and supernatant fractions showed that, in the presence of TF and the cold-shock promoter cspA, the soluble expression of TF-BpFae had increased when compared with the expression using the pET28a-BpFae system. However, enzyme activity assays showed that the soluble protein had negligible enzyme activity (0.04 U mL−1). Although the activity of the soluble enzyme was higher than that of pET28a-Bpfae, considering the content of soluble protein, we speculated that the low activity was because of incorrect protein folding. The presence of the molecular chaperone TF may affect protein function. Enzyme activity was still very low after removal of the fusion tag (data not shown). The low activity suggests that soluble protein production is achieved but a large population of the produced BpFae is misfolded and/or not completely folded. Thus, although the protein adopts a stable soluble conformation, the active site is not in its native state to enable catalysis of the substrate. This phenomenon calls into question the use of protein solubility as an indicator of the desired protein product [Citation24].
Figure 6. SDS-PAGE analysis of the expression of protein TF-BpFae in E. coli BL21(DE3) using pCold-TF vector. M: Molecular weight marker. Lanes 1, 3, 5 and 7: BL21(DE3)-pCold; lanes 2, 4, 6 and 8: BL21(DE3)-pCold-BpFae. Lanes 1 and 2: supernatant of culture medium; lanes 3 and 4: lysate; lanes 5 and 6: supernatant of lysate; lanes 7 and 8: pellet of lysate. The molecular weight of TF-BpFae was predicted as 111 kDa which was consistent with that as seen in the gel.
Expression of pGEX-4T-1-BpFae
The strategy of using protein fusion partners has been effective for increasing expression of target proteins and inhibiting IB formation [Citation67,Citation68]. There are numerous solubility tags reported in the literature; the majority of recent work has focussed on a few tags, notably maltose-binding protein, N-utilization substance A, thioredoxin and GST [Citation68]. The use of GST enhances the production of soluble heterologous proteins remarkably [Citation29,Citation69,Citation70]. The pGEX-4T-1 expression vector uses the Tac promoter, potentially allowing for high levels of expression. The expression of polypeptides in-frame with GST allows for purification of the fusion proteins from crude bacterial extracts under non-denaturing conditions by affinity chromatography on glutathione agarose [Citation71].
The constructed pGEX-4T-1-BpFae fusion plasmid was transformed in E. coli BL21 (DE3) cells and the target fusion protein was overexpressed. However, SDS-PAGE analysis revealed no protein band at the expected molecular weight of ∼85 kDa (). Interestingly, a band around 60 kDa was observed, consistent with the size of the target protein without the fusion GST tag, and the majority of the target protein was expressed in the soluble form. As shown in , the activity of the total protein fraction (0.60 U mL−1) was higher than that of the supernatant (0.40 U mL−1). The insoluble fraction had low activity (0.16 U mL−1), suggesting that aggregation did not disrupt or mask the active site to fully inhibit the activity of the aggregated enzyme. The FAE activity of soluble BpFae expressed by pGEX-4T-1 was 80 and 10 times higher when compared with that of BpFae produced by the pET28a and pCold-TF expression systems, respectively. The supernatants containing BpFae expressed by the three expression plasmids underwent zymogram analysis (). Consistent with enzyme activity analysis, the pGEX-4T-1-BpFae was expressed in the soluble and active form.
Figure 7. Expression of protein GST-BpFae. (a) SDS-PAGE analysis of the expression of protein GST-BpFae in E. coli BL21(DE3) using pGEX-4T-1 vector. M: Molecular weight marker. Lanes 1, 3, 5 and 7: BL21(DE3)- pGEX-4T-1; lanes 2, 4, 6 and 8: BL21(DE3)- pGEX-4T-1-BpFae. Lanes 1 and 2: supernatant of culture medium; lanes 3 and 4: lysate; lanes 5 and 6: supernatant of lysate; lanes 7 and 8: pellet of lysate. The molecular weight of BpFae without GST tag was predicted as 59 kDa which was consistent with that as seen in the gel. (b) Analysis of FAE activity of all protein fractions expressed by pGEX-4T-1.
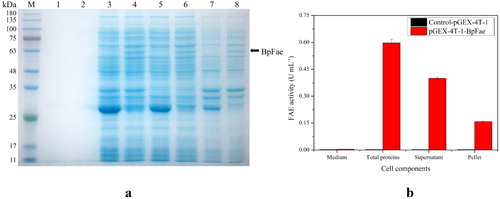
Figure 8. Zymogram analysis of FAEs detected with methyl ferulate as substrates. Samples were supernatant of lysate. 1: pGEX-4T-1-BpFae; 2: pCold-TF-BpFae; 3: pET-28a-BpFae.

The unexpected MW of GST-BpFae observed by SDS-PAGE suggests that the GST fusion tag was cleaved by a hydrolase or another unidentified mechanism. This phenomenon has rarely been observed previously, which has stimulated future interest in exploring why the GST tag was cleaved spontaneously. In future efforts, recombinant BpFae could be purified and verified by N-segment sequencing.
Optimization of feruloyl esterase production
To improve the production of soluble active BpFae, the expression of GST-BpFae in E. coli BL21 (DE3) cells was optimized by examining various culturing and expression parameters (i.e. culture media, initial pH value, inoculum size, IPTG concentration, induction initiation time, induction temperature, rotation speed and post-induction time).
The composition of the medium can substantially affect recombinant protein expression. To select a suitable culture medium for BpFae production, nine commonly used culture media, MX, LBBSMG, TB-GN, LBBM, LB, LBBNM, TB, LBBNG and SOB, were compared. As shown in , GST-BpFae fad maximum activity (0.47 U mL−1) in SOB media, which differed to our previous study, where lactose was used to induce overexpression (the best medium was LB medium) [Citation9]. Although lactose has similar efficacy in inducing E. coli to express heterologous proteins, the results showed that different levels of FAE overexpression were observed when using lactose and IPTG. This may be because IPTG is just an inducer, whereas lactose is both an inducer and a carbon source for fermentation. Therefore, SOB medium was chosen to further study BpFae overexpression.
Figure 9. Effect of media (a), IPTG concentrations (b), induction temperatures (c) and induction times (d) on the production of BpFae.0
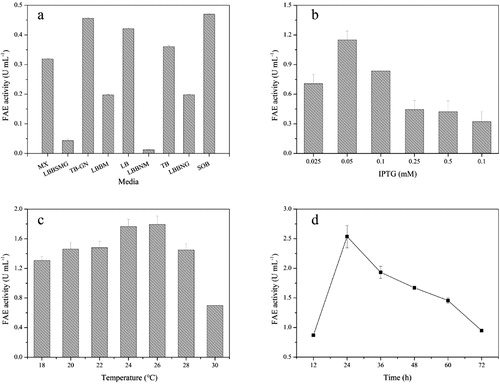
The effect of IPTG concentration on BpFae production and activity was evaluated. As shown in , increasing the IPTG concentration from 0.025 to 0.05 mmol L−1 caused an increase in activity with a maximum of 1.15 U mL−1 obtained. However, the activity decreased dramatically at higher concentrations than 0.05 mmol L−1 IPTG. Hence, 0.05 mmol L−1 IPTG was selected for overexpression. Levels of soluble protein and IBs have been reported to vary significantly with increasing concentrations of IPTG [Citation72]. Induction with high concentrations of IPTG can cause folding and transportation of recombinant proteins to fail because the rate of expression is higher than the rate of folding, resulting in aggregation of most of the target proteins into IBs. Moreover, high concentrations of IPTG are known to be toxic to E. coli [Citation73]. Thus, lower concentrations of the inducer should have a positive effect on the solubility and proper folding of proteins. This activity (1.15 U mL−1) is lower than the result using the optimal lactose concentration (i.e. 2.31 U mL−1) [Citation9]. That may be because the biomass and the content of BpFae in the supernatant after cell disruption (the content of soluble protein) were lower when using IPTG rather than lactose as the inducer, and there were a few inclusion bodies when using IPTG to induce overexpression (there were no inclusion bodies when lactose was used as the inducer) [Citation9].
The effect of induction temperatures on FAE activity was investigated (). Lower temperatures of 18–22 °C reduced the cell growth rate and yield, whereas higher temperatures of 28 and 30 °C were not suitable for protein expression because these temperatures facilitated aggregation of the target protein. Hence, 26 °C was selected for overexpression with 1.79 U mL−1 being the highest activity obtained, which is different from the result (28 °C) of using lactose as a inducer [Citation9]. The best temperature for post-induction is often when the induction rate and cell growth rate are balanced, which means different inducers matching different optimal temperatures. Other expression conditions including the initial pH of the SOB medium (5.0), induction initiation time (4 h), inoculum size (0.8%, v/v) and rotation speed (240 rpm) were also optimized. The effect of induction time on FAE activity of BpFae was then determined. As shown in , increasing the post-induction time from 12 to 24 h caused an increase in activity with a maximum of 2.54 U mL−1. The activity decreased dramatically once cells were grown for more than 24 h post-induction.
Under the optimized expression conditions (i.e. a SOB medium with an initial pH of 5.0, 0.8% (v/v) inoculum volume percentage, induction initiation time of 4 h, IPTG concentration of 0.05 mmol L−1, an induction temperature of 26 °C, shaking at 240 rpm and a post-induction incubation period of 24 h) the FAE activity (2.54 U mL−1) of BpFae increased over 6-fold when compared with the FAE activity before optimization. The FAE activity in this study is consistent with that of TtFAEB1 (2.52 U mL−1) from Thielavia terrestris expressed by Pichia pastoris [Citation74], but is much higher than that of FAE AN1772.2 (0.0045 U mL−1) from Aspergillus nidulans expressed by Saccharomyces cerevisiae [Citation43]. And, another research conducted by our group showed that lactose is a better inducer than IPTG for BpFae production with the FAE activity of 7.43 U mL−1 [Citation9].
Conclusions
In this study, a novel feruloyl esterase BpFae from B. pyrrocinia B1213 was successfully expressed using the pGEX-4T-1 vector in E. coli BL21 (DE3) cells. The recombinant BpFae using this vector expression system was produced in the soluble, active form. Analysis of the primary sequence and 3-D modelling of BpFae using bioinformatics tools revealed that BpFae is a unique and promising candidate for further studies of FAEs. The production level of recombinant BpFae was sixfold higher following optimization of the overexpression conditions. Considering the uniqueness of BpFae, future efforts will focus on investigating its biochemical features, including the mechanism of catalysis, and its potential for industrial applications.
Author contributions
GF and XL conceived and designed the study. ZF, YZ, CT, HL, QL and RY performed the experiments. GF performed data analyses and wrote the paper. XL reviewed and edited the manuscript. All authors have read and approved the final manuscript.
Acknowledgment
We thank Andrew Dingley for insightful discussions and providing language help.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Schulz K, Nieter A, Scheu AK, et al. A type D ferulic acid esterase from Streptomyces werraensis affects the volume of wheat dough pastries. Appl Microbiol Biotechnol. 2018;102(3):1269–1279.
- Topakas E, Vafiadi C, Christakopoulos P. Microbial production, characterization and applications of feruloyl esterases. Process Biochem. 2007;42(4):497–509.
- Dilokpimol A, Makela MR, Aguilar-Pontes MV, et al. Diversity of fungal feruloyl esterases: updated phylogenetic classification, properties, and industrial applications. Biotechnol Biofuels. 2016;9:231.
- Record E, Asther M, Sigoillot C, et al. Overproduction of the Aspergillus niger feruloyl esterase for pulp bleaching application. Appl Microbiol Biotechnol. 2003;62(4):349–355.
- Zduńska K, Dana A, Kolodziejczak A, et al. Antioxidant properties of ferulic acid and its possible application. Skin Pharmacol Physiol. 2018;31(6):332–336.
- Ou S, Kwok K. Ferulic acid: pharmaceutical functions, preparation and applications in foods. J Sci Food Agric. 2004;84(11):1261–1269.
- Oliveira DM, Mota TR, Oliva B, et al. Feruloyl esterases: biocatalysts to overcome biomass recalcitrance and for the production of bioactive compounds. Bioresour Technol. 2019;278:408–423.
- Xu Z, He H, Zhang S, et al. Characterization of feruloyl esterases produced by the Four Lactobacillus species: L. amylovorus, L. acidophilus, L. farciminis and L. fermentum, isolated from ensiled corn stover. Front Microbiol. 2017;8:941.
- Fan G, Zhu Y, Fu Z, et al. Optimization of fermentation conditions for the production of recombinant feruloyl esterase from Burkholderia pyrrocinia B1213. 3 Biotech. 2020;10(5):216.
- Goldstone DC, Villas-Boas SG, Till M, et al. Structural and functional characterization of a promiscuous feruloyl esterase (Est1E) from the rumen bacterium Butyrivibrio proteoclasticus. Proteins. 2010;78(6):1457–1469.
- Cai S, Li J, Hu F, et al. Cellulosilyticum ruminicola, a newly described rumen bacterium that possesses redundant fibrolytic-protein-encoding genes and degrades lignocellulose with multiple carbohydrate- borne fibrolytic enzymes. Appl Environ Microbiol. 2010;76(12):3818–3824.
- Lai KK, Lorca GL, Gonzalez CF. Biochemical properties of two cinnamoyl esterases purified from a Lactobacillus johnsonii strain isolated from stool samples of diabetes-resistant rats. Appl Environ Microbiol. 2009;75(15):5018–5024.
- Uraji M, Arima J, Inoue Y, et al. Application of two newly identified and characterized feruloyl esterases from Streptomyces sp. in the enzymatic production of ferulic acid from agricultural biomass. PLoS One. 2014;9(8):e104584.
- Hunt CJ, Tanksale A, Haritos VS. Biochemical characterization of a halotolerant feruloyl esterase from Actinomyces spp.: refolding and activity following thermal deactivation. Appl Microbiol Biotechnol. 2016;100(4):1777–1787.
- Blum DL, Kataeva IA, Li X, et al. Feruloyl esterase activity of the Clostridium thermocellum cellulosome can be attributed to previously unknown domains of XynY and XynZ. J Bacteriol. 2000;182(5):1346–1351.
- Sal FA, Colak DN, Guler HI, et al. Biochemical characterization of a novel thermostable feruloyl esterase from Geobacillus thermoglucosidasius DSM 2542T). Mol Biol Rep. 2019;46(4):4385–4395.
- Hassan S, Hugouvieux-Cotte-Pattat N. Identification of two feruloyl esterases in Dickeya dadantii 3937 and induction of the major feruloyl esterase and of pectate lyases by ferulic acid. J Bacteriol. 2011;193(4):963–970.
- Crepin VF, Faulds CB, Connerton IF. Functional classification of the microbial feruloyl esterases. Appl Microbiol Biotechnol. 2004;63(6):647–652.
- Li J, Shen W, Fan G, et al. Screening, purification and characterization of lipase from Burkholderia pyrrocinia B1213. 3 Biotech. 2018;8(9):387.
- Li J, Zhang J, Yadav MP, et al. Biodegradability and biodegradation pathway of di-(2-ethylhexyl) phthalate by Burkholderia pyrrocinia B1213. Chemosphere. 2019;225:443–450.
- Donaghy J, Kelly PF, McKay AM. Detection of ferulic acid esterase production by Bacillus spp. and lactobacilli. Appl Microbiol Biotechnol. 1998;50(2):257–260.
- Rumbold K, Biely P, Mastihubova M, et al. Purification and properties of a feruloyl esterase involved in lignocellulose degradation by Aureobasidium pullulans. Appl Environ Microbiol. 2003;69(9):5622–5626.
- Yang S, Tang L, Yan Q, et al. Biochemical characteristics and gene cloning of a novel thermostable feruloyl esterase from Chaetomium sp. J Mol Catal B Enzym. 2013;97:328–336.
- Rosano GL, Ceccarelli EA. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol. 2014;5:172–2019.
- Saini P, Wani SI, Kumar R, et al. Trigger factor assisted folding of the recombinant epoxide hydrolases identified from C. pelagibacter and S. nassauensis. Protein Expr Purif. 2014;104:71–84.
- Kim EK, Moon JC, Lee JM, et al. Large-scale production of soluble recombinant amyloid-β peptide 1-42 using cold-inducible expression system . Protein Expr Purif. 2012;86(1):53–57.
- Hammarstrom M, Hellgren N, Berg S, et al. Rapid screening for improved solubility of small human proteins produced as fusion proteins in Escherichia coli. Protein Sci. 2002;11(2):313–321.
- Zhao X, Li G, Liang S. Several affinity tags commonly used in chromatographic purification. J Anal Methods Chem. 2013;2013:581093.
- Yu H, Ma Q, Lin J, et al. Expression and purification of GST-FHL2 fusion protein. Genet Mol Res. 2013;12(4):6372–6378.
- Rabhi-Essafi I, Sadok A, Khalaf N, et al. A strategy for high-level expression of soluble and functional human interferon alpha as a GST-fusion protein in E. coli. Protein Eng Des Sel. 2007;20(5):201–209.
- Kumar S, Stecher G, Li M, et al. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549.
- Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42(Web Server issue):W320–W324.
- Palm W32 GJ, Reisky L, Bottcher D, et al. Structure of the plastic-degrading Ideonella sakaiensis MHETase bound to a substrate. Nat Commun. 2019;10(1):1717.
- Xu Z, Wang T, Zhang S. Extracellular secretion of feruloyl esterase derived from Lactobacillus crispatus in Escherichia coli and its application for ferulic acid production. Bioresour Technol. 2019;288:121526.
- Antonopoulou I, Papadopoulou A, Iancu L, et al. Optimization of enzymatic synthesis of l-arabinose ferulate catalyzed by feruloyl esterases from Myceliophthora thermophila in detergentless microemulsions and assessment of its antioxidant and cytotoxicity activities. Process Biochem. 2018;65:100–108.
- Laemmli U. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685.
- Camacho-Ruiz MA, Camacho-Ruiz RM, Armendariz M, et al. Corn bran as potential substrate for high production of feruloyl and acetylxylan esterases by solid state fermentation. Rev Mex Ing Quim. 2016;15(1):11–21.
- Golotin VA, Balabanova LA, Noskova YA, et al. Optimization of cold-adapted alpha-galactosidase expression in Escherichia coli. Protein Expr Purif. 2016;123:14–18.
- Chen Y, Huang W, Zhou X, et al. Medium optimization for β-1, 3-1, 4-glucanase production by recombinant Escherichia coli. J Xiamen Univ Nat Sci. 2011;50(5):896–902.
- Zamani M, Berenjian A, Hemmati S, et al. Cloning, expression, and purification of a synthetic human growth hormone in Escherichia coli using response surface methodology. Mol Biotechnol. 2015;57(3):241–250.
- Wu M, Abokitse K, Grosse S, et al. New feruloyl esterases to access phenolic acids from grass biomass. Appl Biochem Biotechnol. 2012;168(1):129–143.
- Koseki T, Hori A, Seki S, et al. Characterization of two distinct feruloyl esterases, AoFaeB and AoFaeC, from Aspergillus oryzae. Appl Microbiol Biotechnol. 2009;83(4):689–696.
- Shin H, Chen R. A type B feruloyl esterase from Aspergillus nidulans with broad pH applicability. Appl Microbiol Biotechnol. 2007;73(6):1323–1330.
- Garcia-Conesa MT, Crepin VF, Goldson AJ, et al. The feruloyl esterase system of Talaromyces stipitatus: production of three discrete feruloyl esterases, including a novel enzyme, TsFaeC, with a broad substrate specificity. J Biotechnol. 2004;108(3):227–241.
- Suzuki K, Hori A, Kawamoto K, et al. Crystal structure of a feruloyl esterase belonging to the tannase family: a disulfide bond near a catalytic triad. Proteins. 2014;82(10):2857–2867.
- Vries R, Michelsen B, Poulsen C, et al. The faeA genes from Aspergillus niger and Aspergillus tubingensis encode ferulic acid esterases involved in degradation of complex cell wall polysaccharides. Appl Environ Microbiol. 1997;63(12):4638–4644.
- Fazary AE, Ismadji S, Ju YH. Studies on temperature dependent kinetics of Aspergillus awamori feruloyl esterase in water solutions. Kinet Catal. 2010;51(1):31–37.
- Kroon PA, Williamson G, Fish NM, et al. A modular esterase from Penicillium funiculosum which releases ferulic acid from plant cell walls and binds crystalline cellulose contains a carbohydrate binding module. Eur J Biochem. 2000;267(23):6740–6750.
- Crepin VF, Faulds CB, Connerton IF. A non-modular type B feruloyl esterase from Neurospora crassa exhibits concentration-dependent substrate inhibition. Biochem J. 2003;370(Pt 2):417–427.
- Topakas E, Moukouli M, Dimarogona M, et al. Expression, characterization and structural modelling of a feruloyl esterase from the thermophilic fungus Myceliophthora thermophila. Appl Microbiol Biotechnol. 2012;94(2):399–411.
- Damasio AR, Braga CM, Brenelli LB, et al. Biomass-to-bio-products application of feruloyl esterase from Aspergillus clavatus. Appl Microbiol Biotechnol. 2013;97(15):6759–6767.
- Kuhnel S, Pouvreau L, Appeldoorn MM, et al. The ferulic acid esterases of Chrysosporium lucknowense C1: purification, characterization and their potential application in biorefinery. Enzyme Microb Technol. 2012;50(1):77–85.
- Crepin VF, Faulds CB, Connerton IF. Identification of a type-D feruloyl esterase from Neurospora crassa. Appl Microbiol Biotechnol. 2004;63(5):567–570.
- Yan J, Wang G, Du P, et al. High-level expression and purification of Escherichia coli oligopeptidase B. Protein Expr Purif. 2006;47(2):645–650.
- Chen A, Li Y, Liu X, et al. Soluble expression of pullulanase from Bacillus acidopullulyticus in Escherichia coli by tightly controlling basal expression. J Ind Microbiol Biotechnol. 2014;41(12):1803–1810.
- Jhamb K, Sahoo DK. Production of soluble recombinant proteins in Escherichia coli: effects of process conditions and chaperone co-expression on cell growth and production of xylanase. Bioresour Technol. 2012;123:135–143.
- Ellis J. Proteins as molecular chaperones. Nature. 1987;328(6129):378–379.
- Ben-Zvi AP, Goloubinoff P. Review: mechanisms of disaggregation and refolding of stable protein aggregates by molecular chaperones. J Struct Biol. 2001;135(2):84–93.
- Ulrich HF, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295(5561):1852–1858.
- Tokuriki N, Tawfik DS. Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature. 2009;459(7247):668–673.
- Peng S, Chu Z, Lu J, et al. Co-expression of chaperones from P. furiosus enhanced the soluble expression of the recombinant hyperthermophilic α-amylase in E. coli . Cell Stress Chaperones. 2016;21(3):477–484.
- Cui S, Lin X, Shen J. Effects of co-expression of molecular chaperones on heterologous soluble expression of the cold-active lipase Lip-948. Protein Expr Purif. 2011;77(2):166–172.
- Tong Y, Feng S, Xin Y, et al. Enhancement of soluble expression of codon-optimized Thermomicrobium roseum sarcosine oxidase in Escherichia coli via chaperone co-expression. J Biotechnol. 2016;218:75–84.
- Qing G, Ma L, Khorchid A, et al. Cold-shock induced high-yield protein production in Escherichia coli. Nat Biotechnol. 2004;22(7):877–882.
- Hayashi K, Kojima C. pCold-GST vector: a novel cold-shock vector containing GST tag for soluble protein production. Protein Expr Purif. 2008;62(1):120–127.
- Cheng X, Zhao L, Klosterman S, et al. The endochitinase VDECH from Verticillium dahliae inhibits spore germination and activates plant defense responses. Plant Sci. 2017;259:12–23.
- Nguyen TKM, Ki MR, Son RG, et al. The NT11, a novel fusion tag for enhancing protein expression in Escherichia coli. Appl Microbiol Biotechnol. 2019;103(5):2205–2216.
- Esposito D, Chatterjee DK. Enhancement of soluble protein expression through the use of fusion tags. Curr Opin Biotechnol. 2006;17(4):353–358.
- Kim S, Lee SB. Soluble expression of archaeal proteins in Escherichia coli by using fusion-partners. Protein Expr Purif. 2008;62(1):116–119.
- Smith MA, Gonzalez J, Hussain A, et al. Overexpression of soluble recombinant human lysyl oxidase by using solubility tags: effects on activity and solubility. Enzyme Res. 2016;2016:5098985.
- Guan K, Dixon JE. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. J Biochem Anal Stud. 1991;192(2):262–267.
- Zhang Z, Wang D, Xu Y. Soluble expression of mature Rhizopus chinensis lipase in Escherichia coli and enhancement of its ester synthesis activity. Protein Expr Purif. 2019;163:105443.
- Kosinski MJ, Rinas U, Bailey JE. Isopropyl-β-d-thiogalactopyranoside influences the metabolism of Escherichia coli. Appl Microbiol Biotechnol. 1992;36(6):782–784.
- Meng Z, Yang QZ, Wang JZ, et al. Cloning, characterization, and functional expression of a thermostable type B feruloyl esterase from thermophilic Thielavia terrestris. Appl Biochem Biotechnol. 2019;189(4):1304–1317.