
Abstract
Flax (Linum usitatissimum L.) is an important economic crop worldwide. The lignin content of flax stems directly determines the quality of flax fiber, and excessively high lignin content in the fiber leads to pollutant discharge by the textile industry. Flax seeds also contain lignans, a group of compounds that are structurally similar to lignin and highly beneficial for human health. The biosynthesis of lignan and lignin occurs in the same metabolic pathway. To elucidate the metabolic relationship between these two compounds and the regulatory nodes of their metabolic processes, third generation (Iso-Seq) and second generation (Illumina) sequencing technologies were used in this study to obtain the transcriptomes of a pair of flax cultivars with significant differences in lignan content. The results showed that the differentially expressed genes (DEGs) are significantly enriched in the lignin and lignan biosynthesis pathways. Ten genes with significant differences in expression (|Log2 FC|>4) were selected for quantitative real-time polymerase chain reaction (qRT-PCR) analysis. The expression levels of these genes all were significantly different in Shuangya 4 and NEW at different stages of flax seed and stem development. We speculated that these genes were most likely closely related to the lignan and lignin metabolism. We also found that the lignan and lignin content are significantly negatively correlated with each other during flax seed and stem development. The findings of this study provide a scientific basis for the application of biotechnological methods to the development of new high-quality flax cultivars with high lignan and low lignin content.
Introduction
The advent of high-throughput sequencing technologies has made it possible to interpret the genomes of many plants. In addition, genome sequencing and annotation can be used to obtain the most basic genetic information of a species. Since 2005, second-generation sequencing technologies have been widely used in crop studies. When compared to Sanger sequencing, second-generation sequencing technologies are cheaper, faster, and have higher throughput [Citation1]. The third-generation sequencing technologies that have appeared in recent years also use the ‘sequencing by synthesis’ strategy of second-generation sequencing techniques, but the read length of the former greatly exceeds that of the latter. Therefore, third-generation sequencing technologies hold significant advantages over second generation technologies in de novo sequencing and full-length transcript sequencing [Citation2]. Currently, third-generation sequencing technologies have been used to perform full-length transcriptome sequencing on crops like salvia, sugar beet, corn, sorghum, and cotton [Citation3–7]. In addition, the use of multi-omic techniques (i.e. transcriptome sequencing and degradome sequencing) has allowed researchers to gain a clearer understanding of gene function, and to study the mechanisms that underlie gene transcription and regulation; multi-omics approaches are expected to serve as the mainstay of gene function studies in the post-genomics era.
Flax (Linum usitatissimum L.) is an important economic crop [Citation8], as textiles made from flax fibers (linen) are in great demand. However, the linen industry is facing a severe shortage of high-quality flax fibers, and lignin content is a major determinant of flax fiber quality. Firstly, excessively high lignin content causes the fibers to become hard and brittle, which directly affects the quality and textile properties of the fibers. Secondly, lignin content is a limiting factor for degumming processes and textile quality, and the wastewaters produced by the lignin removal process are extremely polluting [Citation9]. Consequently, there is a significant level of interest among researchers worldwide in the use of genetic engineering to reduce the lignin content of flax crops or to alter their composition, in order to address the aforementioned issues from their root cause [Citation10]. In recent years, the research into lignin synthesis has mainly focused on the metabolic pathway of phenylpropanoid compounds and lignin-specific biosynthesis. These studies have shown that the activities of enzymes like phenylalanine ammonia-lyase (PAL), cinnamate 4-hydroxylase (C4H), 4-coumarate-CoA ligase (4CL), cinnamyl alcohol dehydrogenase (CAD) and cinnamoyl-CoA reductase (CCR) are closely related to the total amount of lignin in flax fibers [Citation11]. The reduction of PAL activity in transgenic tobacco decreases its lignin content, but this mutation also affects the normative growth of the plant [Citation12]. The inhibition of C4H activity significantly decreases the lignin content of transgenic tobacco, and the regulation of C4H expression also decreases the syringyl/guaiacyl (S/G) ratio in lignin, thereby altering the lignin composition; however, no signs of abnormal plant growth were observed [Citation13]. Studies on multiple transgenic plants have shown that the function of 4CL is not always the same in different plants, and the results obtained after altering 4CL expression differ from one plant to another [Citation14–16]. Based on the histochemical staining of lignin, a study found that CAD overexpression increased vein and stem lignification in transgenic tobacco, whereas CAD overexpression in antisense-transformed plants reduced xylem lignification [Citation17]. Antisense inhibition of the CCR gene in Arabidopsis thaliana significantly reduced the lignin content in the xylem, and it also dramatically loosened the secondary cell wall of interfascicular fibers and vessels, which resulted in abnormal growth [Citation18].
Flax seeds are extremely valuable products, as research has shown that flaxseed is rich in functional nutrients like α-linolenic acid, flaxseed gum, flaxseed protein and lignans [Citation19]. Flax is an extremely rich source of lignans, as their lignan content is tens or hundreds of times that of other plants [Citation20,Citation21]. For this reason, flax is considered by some as the ‘king of lignans’. Flax lignans have been shown to be effective in aiding the prevention and treatment of several severe diseases, and they may also play important roles in cancer prevention, strengthening the immune system and improving human health [Citation22–26]. Therefore, the development and utilization of flax lignans is an indispensable part of flax seed refinement and utilization, as lignans greatly increase the added value and competitiveness of flax products. Furthermore, the development of flax lignans is also of utmost importance for ensuring that the flax industry will continue to develop sustainably. There are currently several reports about the genes related to lignan biosynthesis in flax plants. Renouard et al. [Citation27] conducted a quantitative polymerase chain reaction (PCR) analysis of LuPLR1 expression during the growth of flax seeds, and found that the expression of LuPLR1 is positively correlated with the accumulation of lignan.
Phenylpropanoids refer to a family of compounds that contain a benzene ring attached to a three-carbon side chain, which includes flavonoids, anthocyanins, coumarins, and lignin precursors; these compounds and their derivatives are important components in secondary plant metabolism [Citation28,Citation29]. The lignin polymers in plants mainly consist of syringyl units (S), guaiacyl units (G) and p-hydroxyphenyl units (H), which are derived from sinapyl alcohol, coniferyl alcohol and p-coumaryl alcohol, respectively; coniferyl alcohol-derived G-lignin is especially prevalent in angiosperms [Citation30]. In addition to G-lignins, research has shown that coniferyl alcohol also forms another product, lignin, after a series of enzymatic reactions [Citation31]. However, to our knowledge, there are no reports in the literature about the relationship between lignin and lignan metabolism, or the application of this relationship to obtain high-quality flax cultivars with low lignin content and high lignan content.
In this study, we performed the first full-length transcriptome analysis on flax plants using third-generation sequencing technologies, and report for the first time the negative correlation between lignin and lignan content of flax plants. Candidate genes that may be related to lignin and lignan metabolism were screened. The use of genetic engineering to cultivate high-quality flax cultivars with low lignin content and high lignan content is an ideal two-pronged solution to the issues currently being faced by the flax industry. In this study, we attempt to elucidate the biosynthetic relationship between flax lignan and lignin and provide new ideas for the regulation of lignin and lignan biosynthesis. In addition, the findings of this study will be informative for the cultivation of high-quality, environmentally friendly flax cultivars with high lignan content and low lignin content.
Materials and methods
Plant materials
Flax varieties of Shuangya 4 and NEW were used as test materials. They were provided by the Institute of Industrial Crops, Heilongjiang Academy of Agricultural Sciences. Shuangya 4 is the main flax cultivar in Heilongjiang province and has strong stress resistance, high seed yield, high lignan content (15.9 mg/g in flaxseed), low lignin (859 μg/g in stem (5 cm long, 5 cm-10 cm above the cotyledon nodes)) and excellent comprehensive characteristics (unpublished data). NEW is a variety with low lignan content (3.25 mg/g) and high lignin content (1508 μg/g). They were sown at the Harbin Experimental Base of Heilongjiang Academy of Agricultural Sciences (45°65′N, 126°68′E), Harbin, China, in April 2018. The average annual rainfall from seeding to harvest was 350.2 mm and the average annual temperature was 4.26 °C. Each cultivar was planted in 10-m lines, with a 20-cm inter-line gap, and each test had ten rows and the area was 20 m2, with three replicates. The field management was the same as the local field management.
PacBio sequencing
Root, stem, leaf, flower petal, anther and stigma samples were acquired from the Shuangya 4 flax cultivar when it began to flower. RNA from each sample was extracted and equally mixed, and the SMARTerTM PCR cDNA Synthesis Kit was used to synthesize full-length cDNA from the mRNA. The BluePippin size-selection system was used to select full-length cDNA fragments, which were used to construct cDNA libraries of varying size. The selected full-length cDNAs were PCR amplified, and end repair was performed on the full-length cDNAs, followed by the ligation of SMRTbell adaptors and exonuclease digestion. Secondary screening was performed using the BluePippin system to construct the sequencing libraries, which were quantified using a Qubit 2.0 fluorimeter. An Agilent 2100 Bioanalyzer was used to inspect the size of the sequencing libraries. Sequencing was performed only after the sizes of the sequencing libraries matched the expected sizes. Sequencing was performed on a Pacific Bioscience (PacBio) RS II sequencer with eight cells per RNA strand, with a movie time of 240 min.
Raw SMRT sequences were deposited in the National Center for Biotechnology Information (NCBI) and can be accessed in the database (https://www.ncbi.nlm.nih.gov/) under accession PRJNA478803.
Alternative polyadenylation site analysis
The TAPIS pipeline [Citation5] was used to identify the alternative polyadenylation sites of the full-length transcripts. In nucleotide sequences that contain eight adenylates and a maximum of two non-adenosines in the 3′ end, each starting base was identified as an alternative polyadenylation site. The alternative polyadenylation sites of the genome were then extracted by comparing the transcripts to the reference genome.
RNA-seq and differential gene functional annotation
The flax seeds of Shuangya 4 and NEW flax cultivars were obtained 10, 20 and 30 days after flowering, with three biological replicates for each sample. RNA extraction was conducted on a total of 18 samples. Second-generation transcriptome sequencing was performed using an Illumina HiSeq sequencing system. The detailed process of transcriptome analysis was done as described in our team’s previous research [Citation32]. DESeq was used to screen for mRNA DEGs (FDR ≤ 0.005 and normalized fold change ≥ 1) in Shuangya 4 and NEW flax cultivars, which were subsequently subjected to functional annotation and screening (selection criteria: fold change > 2, FDR < 0.01). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed on the mRNA DEGs. Gene ontology (GO) annotation and clustering were also performed on the DEGs to reveal their functions. Raw Illumina sequences were deposited in the National Center for Biotechnology Information (NCBI) and can be accessed in the database (https://www.ncbi.nlm.nih.gov/) under accession PRJNA476039.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Fresh seed and stem (5 cm long, 5 cm–10 cm above the cotyledon nodes) samples at three different developmental stages of 10, 20 and 30 days after flowering were collected. The samples were put into liquid nitrogen immediately and stored at −80 °C until RNA extraction. The method of quantitative real-time PCR (qRT-PCR) was performed as described in our previous report [Citation33]. The sequences of the specific primers are listed in . All the qRT-PCR experiments were performed for three biological replicates, and each with three technical repeats under the same conditions.
Table 1. Classification of high-quality reads of insert.
Determination of lignan content
The seeds of Shuangya 4 and NEW flax cultivars were obtained 10, 20 and 30 days after flowering, with three biological replicates prepared for each sample. Fixation was performed at 105 °C for 30 min, and the samples were then heated at 80 °C until they reached a constant weight. The dried and fixed samples were then grounded into a powder and passed through a #100 mesh. The resulting powder was stored in a desiccator until further use.
In this study, the high-performance liquid chromatography (HPLC) procedure of Charlet et al. [Citation34] (with minor modifications: hydrolysis treatment time of 3 h instead of 2.5 h) was used to quantify the lignan content of the flax seeds. HPLC was performed using an Agilent 1260 HPLC and a diode array detector (DAD). The mobile phases were methanol (A) and ultrapure water (B). The chromatography column was an Agilent C18 (4.6 mm × 250 mm × 5 μm), and the temperature of the column was 35 °C. The detection wavelength was 290 nm, and 20 μL of sample was injected into the column.
Determination of lignin content
The lignin content of the experimental materials was determined using the NYT 2337-2013 Agricultural Industry Standard of the People’s Republic of China (Testing Retted Jute/Retted Kenaf Fibre Lignin with Sulfuric Acid Method), with minor modifications. The root, lower stem (5 cm long, 5 cm–10 cm above the cotyledon nodes), upper stem (5 cm long, 5 cm–10 cm below the first branch), and leaves of Shuangya 4 and NEW flax cultivars were obtained 10, 20, and 30 days after flowering. Three biological replicates were prepared for each sample.
Correlation analysis
The correlation analysis between the seed lignan content and tissue lignin content of flax plants in various stages in growth was performed using SAS 8.2 software.
Results and discussion
The full-length transcriptome of flax
To construct a complete set of transcripts, we collected high-quality RNA samples from the root, stem, leaf, petals, anthers and stigma of Shuangya 4 plants during their flowering period, and then mixed the high-quality RNAs of these tissues. Then the mixed RNAs were sequenced: RNA fragments of varying size were selected (1-2 kb, 2-3 kb and 3-6 kb) to construct libraries, and sequencing was performed using a PacBio RS II. A total of 1,202,336 raw sequence reads were obtained, and each cell generated 150,292 reads on average. After quality control was performed, 550,240 high-quality ROIs were obtained, with 213,559, 211,111 and 125,570 ROIs being obtained from the 1-2 kb, 2-3 kb, and 3-6 kb libraries, respectively. As we had hoped, the average length of the data generated by each library was the same as the estimated value (). Between these reads, 239,600 were non-full-length transcripts, while 258,948 were full-length transcripts. In this study, full-length transcripts were identified by the presence of 5′ end primers, 3′ end primers, and 3′ end poly(A) tails in the read sequence. In the subsequent analyses, we excluded reads shorter than 300 bp and chimeric reads ().
Figure 1. Characterization of flax (Linum usitatissimum L.) transcriptome using Iso-Seq. (A) Classification of high-quality reads. (B) Summary of full-length isoform mapping in 1–2 kb, 2–3 kb, and 3–6 kb libraries. This includes multiple mapping, unique mapping, and unmapped isoforms. (C) Box-plots show distribution patterns of transcript length in reference annotation and PacBio Iso-Seq data (p < 0.001).
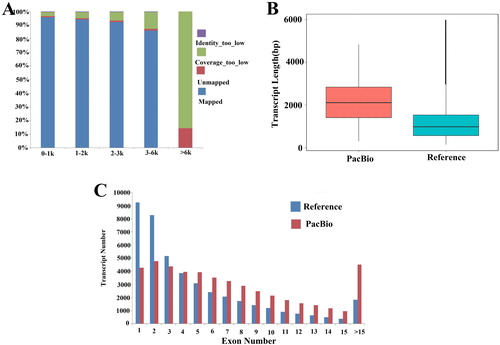
The full-length transcripts were clustered to obtain high-quality non-redundant transcripts. During the preliminary steps of the clustering analysis, it was found that 90% of the transcripts shorter than 6 kb could be uniquely mapped to the genome (). In addition, the full-length transcripts produced by PacBio sequencing (average 2206 bp) were found to be longer than the reference genome (average 1253 bp) (). Furthermore, our analysis discovered that 4258 genes only contain one exon, which is lower than that annotated in the reference genome (9227 genes). In the PacBio data, the number of genes that contain at least 10 exons is higher than that of the reference genome (13,603 versus 6244). The PacBio Iso-Seq technique determined that each gene contains 7.4 exons on average, which is 1.7 times that of the reference genome ().
Flax is an important economic crop, and the findings that have been published about flax genome sequencing have helped to promote basic research about flax plants [Citation35,Citation36]. In the present study, Iso-Seq sequencing technology was used to produce 20.34 Gb of clean data from eight cells, and a total of 113,471 consensus transcript sequences were obtained. After corrections were performed using non-full-length sequences, we obtained 95,403 high-quality full-length transcripts, as well as 18,068 low-quality full-length transcripts. Error correction was then performed on the low-quality sequences using data derived from a second-generation sequencing technique. The gene annotations, isoform transcripts and UTRs of the flax genome were updated using these data. The merging of these data with second-generation sequencing datasets will help to promote functional genomics research on flax plants.
Splicing regions and alternative splicing analysis
One of the most important uses of PacBio Iso-Seq is its ability to compare different full-length transcripts of the same gene to identify alternative splicing events. However, there are no previous studies reporting alternative splicing events in the flax genome. Based on the high-confidence full-length transcripts that were identified in our study, we conducted a systematic analysis of the alternative splicing events that occurred in the flax genome. Firstly, we identified 16,391 alternative splicing events in 9236 genes. Five types of alternative splicing events were identified using the Astalavista [Citation37] program, including exon skipping (ES), alternative 3′ splice sites (A3’S), mutually exclusive exons (ME), alternative 5′ splice sites (A5’S) and intron retention (IR) (). By counting the number of genes corresponding to each type of splicing event, we discovered that IR occurred in 65.53% of the genes. Therefore, IR events account for most of the alternative splicing events in the flax genome (). The second and third most common alternative splicing events are A3’S and A5’S events, respectively. A similar number of A5’S and ES events were observed in this study. At the transcript level, 36,353 new transcripts were identified in 24,564 genes, and 13,758 of these genes contain only one transcript. In the reference genome, most of the genes were annotated as single-transcript genes (43,458 genes); only 13 genes have two transcripts, and no genes have three transcripts. From the PacBio data, there were 5190 genes with a minimum of three transcripts. The PacBio Iso-Seq technique determined that each gene contained 1.23 transcripts on average (), which is 1.2 times that of the reference genome’s annotations. These results indicate that alternative splicing events have increased the complexity of the flax transcriptome.
Figure 2. Classification of AS events and distribution of the number of isoforms per gene. (A) Classification of AS events. images display the AS events: exon skipping (ES), alternative 3′ splice site (A3’S), mutually exclusive exon (ME), alternative 5′ splice site (A5’S), and intron retention (IR). (B) Distribution of the number of isoforms per gene. The average number of isoforms for all genes in reference annotation and PacBio data are shown in blue and red.
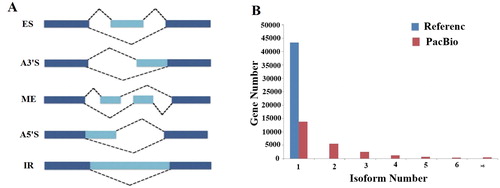
Table 2. Alternative splicing (AS) event statistics.
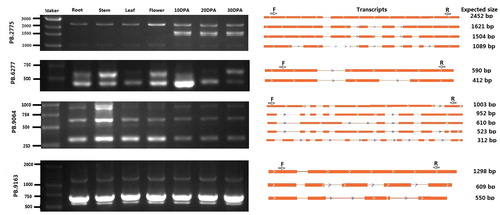
RT-PCR was performed on four randomly selected genes to validate the accuracy of alternative splicing event identification. Firstly, the transcripts of each gene were compared to locate a consensus sequence. Primers were then designed for the sequence, thereby allowing all of the transcripts to be amplified in a single experiment (). The results of this experiment indicate that the sizes of the amplified fragments were consistent with the expected sizes. In addition, some of the transcripts exhibited tissue specificity in their expression. For example, the PB.2775 gene codes for a MATE-like protein, and is able to transcribe seven transcript isoforms through A5’S events. Between these transcripts, at least two are only expressed in flax seeds during their development, and they are not expressed in other parts of the plant ( ). Alternative splicing is an important mechanism to regulate gene expression and proteome diversity, and is an important reason for the large difference in the number of flax genes and proteins. Therefore, the analysis of alternative splicing in the flax genome laid a foundation for the follow-up gene function annotation analysis.
Figure 3. RT-PCR validation of AS events for four genes.
Note: Gel bands in each figure show DNA markers and PCR results in seven samples (DPA, days post-anthesis). The transcript structure of each isoform is shown in the right panel. Yellow boxes indicate exons and lines with arrows indicate introns. PCR primers (F, forward and R, reverse) are shown on the first isoform of each gene. The length of each full-length isoform is shown after the transcript structure. (In order to facilitate understanding, we have cut the gel picture accordingly. Please refer to the original image in )
The analysis of alternative splicing events performed in our study will accelerate the development of functional genomics research with respect to flax plants in the future. Owing to the lack of an accurately annotated flax transcriptome data, functional differences between transcripts of the same gene have not been noted in previous flax plant studies. In eukaryotic lifeforms, alternative splicing often leads to the generation of different transcripts. Based on Iso-Seq data, we have identified 16,391 alternative splicing events in 9236 genes, with IR events accounting for the vast majority of these events. This result is consistent with the findings of previous studies in other plants [Citation7,Citation38–40]. By analyzing the transcripts of various tissues, we have found that alternative splicing may have resulted in the same gene with different functions across different tissues. These results generally indicate that alternative splicing events have greatly increased the complexity of the flax transcriptome, and they also remind us that transcript-level approaches are necessary in gene studies.
Differential gene enrichment analysis and functional annotation
The mRNA expression levels of Shuangya 4 and NEW cultivars varied significantly during their development in the post-flowering stage. A total of 1643 DEGs was identified, of which 678 were upregulated, while 965 were downregulated. We observed 965 DEGs 10 days after flowering (the highest number of DEGs, by far), with 432 being upregulated and 533 being downregulated; 404 DEGs were observed 30 days after flowering, and a mere 274 DEGs were observed 20 days after flowering. Functional annotation was performed on the 41 DEGs with |fold change| > 3 that were detected in two or more developmental stages. Seven DEGs were annotated as UDP-glycosyltransferase. In addition, three DEGs were annotated as laccase, peroxidase and cytochrome P450, respectively.
In addition, five DEGs were annotated in all three developmental periods (). KEGG pathway analysis found that the functions of the ‘10 days after flowering’ (henceforth denoted the ‘10-day’ stage) DEGs in both cultivars were mainly related to plant-pathogen interactions, peroxisome, protein processing in the endoplasmic reticulum, spliceosomes and ribosomes ((A)). The functions of the ‘20 days after flowering’ (20-day stage) DEGs were mainly related to phenylpropanoid metabolism, spliceosomes, plant hormone signal transduction, ribosomes, and starch and sucrose metabolism ((B)). The functions of the ‘30 days after flowering’ (30-day stage) DEGs pertained to phenylpropanoid metabolism, starch and sucrose metabolism, photosynthesis-antenna proteins, DNA replication and photosynthesis ((C)). In particular, the raw materials required in all stages of plant growth and development are supplied by starch and sucrose metabolism [Citation41], whereas spliceosomes and ribosomes are large and complex molecular machines that are indispensable for eukaryotic gene expression. We also identified DEGs that participate in photosynthetic antenna proteins and photosynthesis-related pathways, which hints that these DEGs may play a role in the formation and accumulation of flaxseed nutrients. More importantly, it was shown that many DEGs participate in phenylpropanoid metabolism, and these DEGs generally result in significant phenylpropanoid enrichment at the 20-day and 30-day stages. Hence, the phenylpropanoid metabolism pathway could produce large amounts of secondary metabolites [Citation42,Citation43].
Table 3. Correlation analysis between seed lignan content and tissue lignin content of flax plants in various stages of growth.
Upon further analysis, it was discovered that the DEG functions of the Shuangya 4 and NEW cultivars exhibit significant enrichment in the (+)-pinoresinol catabolic process, (-)-lariciresinol biosynthetic process, (+)-lariciresinol biosynthetic process, protein-chromophore linkage, peptidyl-histidine phosphorylation, amine transport and cellular responses to cold (D–F)). In addition to the pathways and processes necessary for plant growth, it was revealed by KEGG pathway analysis and GO annotation that many of the DEGs participate in the phenylpropanoid metabolism pathway in the KEGG pathway, as well as the (+)-pinoresinol catabolic process, (-)-lariciresinol biosynthetic process, and (+)-lariciresinol biosynthetic process in the GO annotations (). Phenylpropanoid metabolism is a part of the lignin metabolic pathway in plants [Citation44], whereas the (+)-pinoresinol catabolic process, (-)-lariciresinol biosynthetic process, and (+)-lariciresinol biosynthetic process are necessary steps for lignan biosynthesis in flax plants [Citation45]. Therefore, we concluded that these DEGs play an important role in the lignin and lignan metabolism of flax plants.
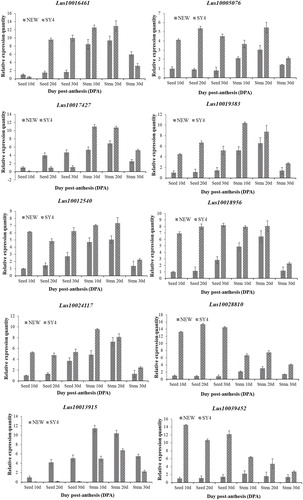
Figure 4. Expression patterns of UGTs in Shuangya 4 and NEW in each stage of flaxseed and stem development.
Note: The total RNA of the flax plants was obtained at 0–30 days after flowering to facilitate an analysis of the expression patterns of each UGT gene in each stage of flaxseed and stem development. The quantitative PCR data are the mean of three biological replicates, whereas the bars represent the standard deviations.
In the transcriptome data of flaxseeds that were collected from these flax cultivars at different stages of growth, significant DEG enrichment was observed in the KEGG phenylpropanoid metabolism pathway. The derivatives of phenylpropanoid are p-coumaryl alcohol, coniferyl alcohol and sinapyl alcohol, which serve as substrates for the biosynthesis of G-lignin, H-lignin and S-lignin production, respectively [Citation46]. In particular, coniferyl alcohol also serves as the substrate for lignan production. As the DEGs are significantly enriched in the phenylpropanoid metabolism pathway, it is implied that these DEGs are closely related to lignin and lignan metabolism. In the GO annotations, the DEGs are mainly enriched in the (+)-pinoresinol catabolic process, (-)-lariciresinol biosynthetic process and (+)-lariciresinol biosynthetic process, which are necessary processes for lignan biosynthesis [Citation27]. These results imply that the genes related to lignin and lignan metabolism are strongly coupled to each other.
Differentially expressed genes were verified by qRT-PCR
We selected ten genes (|Log2FC|>4) for qRT-PCR validation (). These genes included lus10016461 (UDP-glycosyltransferase 1), lus10005076 (UDP-glycosyltransferase 1), lus10024117 (UDP-glycosyltransferase 1), lus10012540 (Peroxidase), lus10013915 (Pheophorbide a oxygenase), lus10017427(Laccase), lus10018956 (Sugar transporter ERD6-like), lus10019383 (Tousled-like kinase), lus10028810 (ATP-dependent transporter, putative) and lus10039452 (Cellular repressor of stimulated genes). The expression quantity of these ten genes at different stages of flax seed and stem development in different flax varieties were tested by qRT-PCR ( ). We found that except for lus10017427 and lus10013915, the expression levels of the other genes in Shuangya 4 were significantly higher than those in NEW at different stages of flax seed and stem development. On the contrary, the expression levels of lus10013915 in NEW was significantly higher than that in Shuangya 4 at different stages of flax seed and stem development. The expression levels of lus10017427 in NEW was significantly higher than that in Shuangya 4 at different stages of flax seed, but the opposite expression pattern appeared at different stages of stem development. In addition, it was found that the expression patterns of these genes at each stage of flax seed development were basically consistent with the transcriptome sequencing results (), which proved the reliability of the results from this study.
Three genes were annotated as the UDP-glycosyltransferase 1 family (UGTs), implying that these play a role in the glycosylation of secondary metabolites like lignan or lignin. Lin et al. [Citation47] created a ugt72b1 mutant in Arabidopsis thaliana, which resulted in severe ectopic lignification and the enhancement of cell wall lignification; lignin content analyses on this mutant also revealed significantly higher levels of lignin than the wildtype. In addition, Lin et al. [Citation47] also measured the p-coumaryl alcohol, coniferyl alcohol and sinapyl alcohol (i.e. lignin precursor) content of the mutant. It was determined that the coniferyl alcohol content of the mutant greatly exceeds that of the wildtype and even surpasses that of the overexpression line; this observation greatly puzzled the authors. Our research has resolved this puzzle, as we have found that lignan and lignin compete for the same substrate-coniferyl alcohol. Furthermore, UGTs are regulatory hub genes in lignan and lignin metabolism. Therefore, we speculate that the function of the UGTs was disrupted in the mutant strain, which blocked or weakened the pathway that converts coniferyl alcohol into lignan, thus resulting in large amounts of coniferyl alcohol being accumulated within the mutant. Our hypothesis also finds support from the results of previous studies, albeit from a different viewpoint. A preliminary study by Ghose et al. [Citation48] discovered that the UGT74S1 gene plays a role in the glycosylation of SECO into lignan, thus indicating that UGTs could play a significant role in flax lignan biosynthesis. Fofana et al. [Citation49] performed EMS mutagenesis on CDC Bethune flax and obtained 1996 M2 mutants; between these mutants, 93 strains exhibited mutations in the exonic region of UGTS74S1, which inhibited lignan synthesis in their M3 and M4 descendants. The other seven genes also showed significant differences at different stages of seed and stem development in the two flax varieties. However, these genes functions have no related reports so far, so we hypothesized that these genes may be closely related to lignin and lignin metabolism. Their specific functions need to be further tested and verified.
Correlation between lignin and lignan content at each stage of development
Although it has been demonstrated that lignin and lignan are both produced by the same metabolic pathway [Citation31], a relationship between lignin and lignan has yet to be reported in the literature. Therefore, studies about the relationship between lignin and lignan will be of significant importance for the cultivation of high-quality flax cultivars with high lignan and low lignin content. A correlation analysis was performed between the seed lignan content and tissue lignin content of flax plants in various stages in growth (). At 10 days after flowering, the lignan content of flax seeds was significantly negatively correlated with lignin content in the roots, lower stem and upper stem, and significantly negatively correlated with lignin content in the leaves. At 10-30 days after flowering, the lignan content of flax seeds was negatively correlated with the lignin content of the roots, but this was not significant. Negative correlations were observed between seed lignan content and the lignin content of the lower stem and upper stem, respectively. A positive correlation was observed between seed lignan content and leaf lignin content, albeit at a non-significant level. Therefore, the accumulation of seed lignan and stem lignin in the post-flowering period may have competed for the same substrate, which resulted in a negative correlation between seed lignan content and stem lignin content in flax plants.
The processing and utilization of plant materials are mainly limited by the presence of lignin. Excessively high levels of lignin content in plant fibers will result in hard and brittle fibers, which are difficult to weave into textiles. In addition, due to the immense difficulty of lignin removal, plant materials with high lignin content will result in high degumming costs and industrial pollutant discharges [Citation50]. Therefore, the lignin content of a flax plant will directly affect the quality of its fibers and thus, its economic value. Flax seeds also contain lignan, which is a functional component [Citation19]; the development and utilization of this functional component will greatly increase the added value of flax products. However, studies on flax plants have generally focused either on lignan or lignin, and there have been no reports about the relationship between these compounds. Huis et al. [Citation31] utilized a multi-omics approach that combined metabolomics and transcriptomics to study lignification in the woody part of flax stems and extraxylary bast fibers. In their study, they briefly mentioned that coniferyl alcohol, a metabolic intermediate, could ultimately be converted into lignin and lignan. However, there was no mention of the derivative relations between lignin and lignan. By measuring the seed lignan content and stem lignin content of flax plants in various stages of the post-flowering period, we observed that the lignan and lignin content are negatively correlated with each other.
Conclusions
Full-length transcriptome analysis of flax was performed for the first time in this study, which has helped to fill gaps in the flax reference genome. We discovered that alternative splicing events have increased the complexity of the flax transcriptome. A concerted analysis based on second-generation sequencing revealed that DEGs are mainly enriched in the phenylpropanoid metabolism pathway, which is the pathway that produces lignan and lignin. Based on the measurement of stem lignin content and flaxseed lignan content in different stages of plant growth on flax cultivars with significant differences in lignan content, we found that the lignan content of a flax plant is negatively correlated with its lignin content. Many of the DEGs were functionally annotated as UGTs. Based on the results of previous studies, we speculate that UGTs are regulatory node genes for lignan and lignin metabolism.
Supplemental Material
Download PDF (144.8 KB)Supplemental Material
Download PDF (55 KB)Supplemental Material
Download PDF (60.2 KB)Supplemental Material
Download PDF (1.3 MB)Supplemental Material
Download PDF (175.2 KB)Supplemental Material
Download PDF (751.8 KB)Supplemental Material
Download PDF (167.1 KB)Acknowledgement
The authors would like to thank the National Bast Fiber Crops Germplasm Improvement Center of Flax Branch Center for kindly supplying the experimental platform.
Disclosure statement
No potential conflict of interest was reported by the authors.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files. Raw Illumina sequences were deposited in the National Center for Biotechnology Information (NCBI) and can be accessed in the database (http://www.ncbi.nlm.nih.gov/biosample/7551536) under accession PRJNA478803 and PRJNA476039.
Additional information
Funding
Related Research Data
References
- Shendure J, Ji HL. Next-generation DNA sequencing. Nat Biotechnol. 2008;26(10):1135–1145.
- Manrao EA, Derrington IM, Laszlo AH, et al. Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nat Biotechnol. 2012;30(4):349-U174.
- Xu Z, Peters RJ, Weirather J, et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015;82(6):951–961.
- Minoche AE, Dohm JC, Schneider J, et al. Exploiting single-molecule transcript sequencing for eukaryotic gene prediction. Genome Biol. 2015;16(1):184.
- Abdel-Ghany SE, Hamilton M, Jacobi JL, et al. A survey of the Sorghum transcriptome using single-molecule long reads. Nat Commun. 2016;7:1–11.
- Wang B, Tseng E, Regulski M, et al. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat Commun. 2016;7:1–13.
- Wang M, Wang P, Liang F, et al. A global survey of alternative splicing in allopolyploid cotton: landscape, complexity and regulation. New Phytol. 2018;217(1):163–178.
- Vaisey-Genser M, Morris DH. Introduction: history of the cultivation and uses of flaxseed. In: Muir AD, Westcott ND, editors. Flax: the genus Linum. Boca Raton (FL): CRC Press; 2003. p. 1–21.
- Magdalena WK, Starzycki M, Zebrowski J, et al. Lignin deficiency in transgenic flax resulted in plants with improved mechanical properties. J Biotechnol. 2007;128:919–934.
- Summerscales J, Dissanayake NPJ, Virk AS, et al. A review of bast fibres and their composites. Part 1-Fibres as reinforcements. Compos A: Appl Sci Manuf. 2010;41(10):1329–1335.
- Chen HY, Babst BA, Nyamdari B, et al. Ectopic expression of a loblolly pine class II 4-coumarate:CoA ligase alters soluble phenylpropanoid metabolism but not lignin biosynthesis in Populus. Plant Cell Physiol. 2014;55(9):1669–1678.
- Elkind Y, Edwards R, Mavandad M, et al. Abnormal plant development and down-regulation of phenylpropanoid biosynthesis in transgenic tobacco containing a heterologous phenylalanine ammonia-lyase gene. Proc Natl Acad Sci USA. 1990;87(22):9057–9061.
- Sewalt V, Ni W, Blount JW, et al. Reduced lignin content and altered lignin composition in transgenic tobacco down-regulated in expression of L-phenylalanine ammonia-lyase or cinnamate 4-hydroxylase. Plant Physiol. 1997;115(1):41–50.
- Lu H, Zeng Q, Zhao YL, et al. Xylem-specific expression of a GRP1.8 promoter: 4CL gene construct in transgenic tobacco. Plant Growth Regul. 2003;41(3):279–286.
- Voelker SL, Lachenbruch B, Meinzer FC, et al. Antisense down-regulation of 4CL expression alters lignification, tree growth, and saccharification potential of field-grown poplar. Plant Physiol. 2010;154(2):874–886.
- Wang Y, Zhang X, Yang S, et al. Heterogenous expression of Pyrus pyrifolia PpCAD2 and PpEXP2 in tobacco impacts lignin accumulation in transgenic plants. Gene. 2017;637:181–189.
- Goujon TFV, Ferret V, Mila I, et al. Down-regulation of the AtCCR1 gene in Arabidopsis thaliana: effects on phenotype, lignins and cell wall degradability. Planta. 2003;217(2):218–228.
- Meagher LP, Beecher GR. Assessment of data on the lignan content of foods. J Food Compos Anal. 2000;13(6):935–947.
- Kiely M, Faughnan M, Wähälä K, et al. Phyto-oestrogen levels in foods: the design and construction of the VENUS database. Br J Nutr. 2003;89(S1):S19–S23.
- Blitz CL, Murphy SP, Au DLM. Adding lignan values to a food composition database. J Food Compos Anal. 2007;20(2):99–105.
- Axelson M, Setchell KDR. The excretion of lignans in rats evidence for an intestinal bacterial source for this new group of compounds. FEBS Lett. 1981;123(2):337–342.
- Alhassane T, Xu XM. Flaxseed lignans: source, biosynthesis, metabolism, antioxidant activity, bio-active components, and health benefits. Compr Rev Food Sci F. 2010;9:261–269.
- Zanwar AA, Hegde MV, Bodhankar SL. Chapter 71-flax lignan in the prevention of atherosclerotic cardiovascular diseases. Polyphenols Hum Health Dis. 2014;2:915–921.
- Kreydin EI, Kim MM, Barrisford GW, et al. Urinary lignans are associated with decreased incontinence in postmenopausal women. Urology. 2015;86(4):716–720.
- Chen FP, Chang CJ, Chao AS, et al. Efficacy of Femarelle for the treatment of climacteric syndrome in postmenopausal women: an open label trial. Taiwan J Obstet Gyne. 2016;55(3):336–340.
- Renouard S, Corbin C, Lopez T, et al. Abscisic acid regulates pinoresinol-lariciresinol reductase gene expression and secoisolariciresinol accumulation in developing flax (Linum usitatissimum L.) seeds. Planta. 2012;235(1):85–98.
- Vogt T. Phenylpropanoid biosynthesis. Mol Plant. 2010;3(1):2–20.
- Gray J, Caparrós-Ruiz D, Grotewold E. Grass phenylpropanoids: regulate before using! Plant Sci. 2012;184:112–120.
- Voxeur A, Wang Y, Sibout R. Lignification: different mechanisms for a versatile polymer. Curr Opin Plant Biol. 2015;23:83–90.
- Huis R, Morreel K, Fliniaux O, et al. Natural hypolignification is associated with extensive oligolignol accumulation in flax stems. Plant Physiol. 2012;158(4):1893–1915.
- Xie D, Dai Z, Yang Z, et al. Genomic variations and association study of agronomic traits in flax. BMC Genom. 2018;19:512.
- Sun J, Xie DW, Zhao HW, et al. Genome-wide identification of the class III aminotransferase gene family in rice and expression analysis under abiotic stress: ATIII gene family in japonica and indica rice. Genes Genom. 2013;35(5):597–608.
- Charlet S, Bensaddek L, Raynaud S, et al. An HPLC procedure for the quantification of anhydrosecoisolariciresinol. Application to the evaluation of flax lignan content. Plant Physiol Bioch. 2002;40(3):225–229.
- Wang Z, Hobson N, Galindo L, et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012;72(3):461–473.
- Zhang JP, Qi YN, Wang LM, et al. Genomic comparison and population diversity analysis provide insights into the domestication and improvement of flax. iScience. 2020;23(4):100967.
- Foissac S, Sammeth M. ASTALAVISTA: dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007;35:297–299.
- Zhang G, Guo G, Hu X, et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010;20(5):646–654.
- Shen Y, Zhou Z, Wang Z, et al. Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell. 2014;26(3):996–1008.
- Thatcher SR, Zhou WG, Leonard A, et al. Genome-wide analysis of alternative splicing in Zea mays: landscape and genetic regulation. Plant Cell. 2014;26(9):3472–3487.
- Vriet C, Smith AM, Wang TL. Root starch reserves are necessary for vigorous re-growth following cutting back in Lotus japonicus. Plos One. 2014;9(1):e87333.
- Shelton D, Leach D, Baverstock P, et al. Isolation of genes involved in secondary metabolism from Melaleuca alternifolia (Cheel) using expressed sequence tags (ESTs). Plant Sci. 2002;162(1):9–15.
- Tian HL, Xu XS, Zhang FS, et al. Analysis of Polygala tenuifolia transcriptome and description of secondary metabolite biosynthetic pathways by Illumina sequencing. Int J Genom. 2015;2015:1–11.
- Barber MS, Mitchell HJ. Regulation of phenylpropanoid metabolism in relation to lignin biosynthesis in plants. Int Rev Cytol. 1997;172:243–293.
- Ayella AK, Trick HN, Wang W. Enhancing lignan biosynthesis by over-expressing pinoresinol lariciresinol reductase in transgenic wheat. Mol Nutr Food Res. 2007;51(12):1518–1526.
- Le Roy J, Blervacq AS, Créach A, et al. Spatial regulation of monolignol biosynthesis and laccase genes control developmental and stress-related lignin in flax. BMC Plant Biol. 2017;17(1):124.
- Lin JS, Huang XX, Li Q, et al. UDP-glycosyltransferase 72B1 catalyzes the glucose conjugation of monolignols and is essential for the normal cell wall lignification in Arabidopsis thaliana. Plant J. 2016;88(1):26–42.
- Ghose K, Selvaraj K, McCallum J, et al. Identification and functional characterization of a flax UDP-glycosyltransferase glucosylating secoisolariciresinol (SECO) into secoisolariciresinol monoglucoside (SMG) and diglucoside (SDG). BMC Plant Biol. 2014;14:82.
- Fofana B, Ghose K, Somalraju A, et al. Induced mutagenesis in UGT74S1 gene leads to stable new flax lines with altered secoisolariciresinol diglucoside (SDG) profiles. Front Plant Sci. 2017;8:1638.
- Vanholme R, Storme V, Vanholme B, et al. A systems biology view of responses to lignin biosynthesis perturbations in Arabidopsis. Plant Cell. 2012;24(9):3506–3529.