
Abstract
The tree Cryptomeria japonica var. sinensis is native to China and is an important forest species widely used for wood production. Here, we sequenced the complete chloroplast (cp) genomes of six wild and six cultivated accessions of this tree. The 12 cp genomes ranged from 131,379 to 131,528 bp. The GC content was 35.4%, similar to other gymnosperm species. The cp genomes lacked typical inverted repeat (IR) regions and encoded 118 genes. Most genes appeared in one copy and 17 genes contained introns. Two multi-copy genes (trnM-CAU × 3, trnQ-UUG × 2) were identified. And 59–61 simple sequence repeats (SSRs) were identified in the whole cp genomes, and most SSR loci consisted of A or T bases. Phylogenetic analysis indicated that wild and cultivated accessions were not clearly differentiated. Our results will provide useful information for the conservation and utilization of this variety.
Supplemental data for this article is available online at https://doi.org/10.1080/13102818.2021.1932592 .
Introduction
The chloroplast (cp) is the most important organelle in green plants as it is the place where photosynthesis and carbon fixation occur. Compared with the nuclear genome, the cp genome is more conserved in terms of gene structure and composition, which is advantageous for the study of taxa at higher taxonomic levels [Citation1, Citation2]. Moreover, the cp genome does not recombine and is uniparentally inherited [Citation3, Citation4], which can elucidate the history and evolution of plant populations [Citation5]. Complete cp genome sequences were first reported for tobacco [Citation6] and liverwort [Citation7] in 1986. In recent years, the rapid progress of next-generation sequencing has enabled us to better understand the molecular and genomic characteristics of cp genomes [Citation8–10].
The cp genomes of higher plants are circular molecules ranging in size from 100 to 200 kb [Citation11]. In angiosperms, the cp genome contains two identical inverted repeat sequences (IRA, IRB) that divide the genome into large (LSC) and small single copy (SSC) regions. It is believed that large IRs can help stabilize the cp genome [Citation12]. The relative size of this typical quadripartite structure remains constant; the gene order and organization are highly conserved [Citation13, Citation14]. However, in gymnosperms, the cp genome of most coniferous species lacks the large IRs, which may lead to more gene loss and structural rearrangement [Citation15–17].
C. japonica var. sinensis, also called Liushan, is a native variety of C. japonica in southeast China. It is one of the most important plantation species widely used for commercial timber. In this study, we sequenced the cp genomes of six wild and six cultivated Liushan trees using a next-generation sequencing platform. We compared the gene content, genome structure, SSRs information, and intraspecific variation among the cp genomes of these 12 accessions. Phylogenetic analysis was also performed to understand the relationship of C. japonica var. sinensis in Cupressaceae family.
Materials and methods
DNA sequencing and genome assembly
Twelve C. japonica var. sinensis accessions, including six wild and six cultivated trees were selected from Tianmu Mountain (119°26’08.83’’E, 30°20’17.17’’N) and Xiapu seed orchard (119°56’13.85’’E, 26°51’58.99’’N) in China, respectively. Fresh leaves of each tree were sampled for total DNA extraction using a modified CTAB protocol [Citation18]. Five micrograms of purified DNA were used to construct the short-insert libraries (average 450 bp) according to the Illumina standard protocol. Genome sequencing was performed using Illumina Hiseq high-throughput sequencing technology. The raw data were filtered to obtain high-quality reads by removing adapters and low-quality sequences using the NGS QC Toolkit v2.3.3 [Citation19]. The complete cp genome of C. japonica (Accession: AP010967) was used as a reference sequence for splicing, assembly and annotation by SPAdes 3.9.0 software [Citation20].
Genome analysis and annotation
To assess the levels of genomic variation between wild and cultivated trees, parsimony informative sites and nucleotide diversity were calculated using DnaSP version 6.1 [Citation21]. The step size was set to 200 bp, with a 600-bp window length. The Dual Organellar GenoMe Annotator (DOGMA) [Citation22] was used for genome annotation based on comparisons of homologous genes with other conifer cp genomes. UGENEORFs finder tool was used to predict open reading frames (ORFs) in the DNA sequences. The tRNA genes were confirmed by tRNAscan-SE version 1.21 [Citation23] with default settings. The rRNA genes were verified using the RNAmmer 1.2 server [Citation24]. A circular gene map of each cp genome was drawn by the online tool of Organellar Genome Draw program (OGDraw) [Citation25].
Phylogenetic analysis
Intraspecific phylogeny analysis was performed using 12 C. japonica var. sinensis and C. japonica base on a data matrix of 82 shared protein-coding genes. Furthermore, interspecific phylogeny analysis of 13 Cupressaceae species was also performed with 71 common protein-coding genes, using Cunninghamia lanceolata as outgroup. The phylogenetic trees were constructed estimated by maximum likelihood (ML) in MEGA X [Citation26]. The bootstrap support of each branch was calculated with 1000 replicates. The bootstrap values are only shown for nodes with greater than 50% support.
Chloroplast SSRs identifying
Simple sequence repeats (SSRs) were detected using the MISA Perl script [Citation27]. The parameter of minimum repeat units was set as 10 for mononucleotide, 6 for dinucleotide and 5 for trinucleotide to hexanucleotide, respectively.
Results and discussion
Chloroplast genome features
The 12 cp genomes of C. japonica var. sinensis ranged from 131,379 to 131,528 bp in length (), with an average GC content of 35.40%, which is the same as C. japonica and similar to Taxodium distichum (35.26%) and Glyptostrobus pensilis (35.31%). Like some species in cupressophytes [Citation28–30], the cp genome lacks the typical quadripartite structure. No large IR region in C. japonica var. sinensis could be detected, which made a distinction into a large (LSC) and small (SSC) single copy region difficult. The complete cp genome contains 118 genes (, Supplemental file 1), including 82 protein-coding genes (69.49%), 32 tRNA genes (27.12%) and four rRNA genes (3.39%). In protein-coding genes, there are 21 genes encoding large and small ribosomal subunits (17.80%), four genes encoding DNA-dependent RNA polymerase (3.39%), 48 genes encoding photosynthesis-related proteins (40.68%), eight genes encoding other proteins (6.78%) and four encoding proteins with unknown functions (3.39%).
Table 1. The cp genomes contents of C. japonica var. sinensis in wild (CFXTM01 to 06) and cultivated trees (CFXP01 to 06).
Table 2. List of genes found in C. japonica var. sinensis cp genome.
Among the total genes in the cp genome, 112 were single-copy genes, and two tRNA genes (trnM-CAU × 3, trnQ-UUG × 2) were multi-copy. Introns can regulate the transcription rate of genes and play an important role in the genes structure and function [Citation31]. A total 17 single-copy genes contained introns (), including 11 protein-coding genes (trnA-UGC’ trnI-AAU’ trnI-AUC’ trnK-AAA’ trnL-UAA’ trnS-CGA’ rps12’ rps16’ rpl2’ rpl16’ rpoC1’ petB’ petD’ atpF’ ndhA’ ndhB and ycf3) and six tRNA genes. In protein-coding genes, rps12 was identified as a trans-spliced gene. The distance between 5’rps12 and 3’rps12 genes was 38.9 kb. The tRNA genes are among the most important versatile molecules responsible for maintaining the protein translation machinery [Citation32, Citation33]. C. japonica var. sinensis has a higher number of tRNAMet and tRNASer genes, and has a lower number of tRNAGly, tRNAIle, tRNAThr and tRNAVal genes (). The tRNAMet species, including initiator tRNAfMet and elongator tRNAMet, is a major player to give rise to other tRNAs [Citation34, Citation35]. We found three copies of tRNAMet in the cp genome, which was different to C. japonica, T. distichum and G. pensilis [Citation28–30]. Four ycf genes (ycf1 to ycf4) have also been identified in the cp genome, but the clpP gene was absent ().
Figure 1. Gene map of C. japonica var. sinensis cp genome. Genes on the outside of the circle are transcribed counter-clockwise, while genes on the inside are transcribed clockwise. Different colours represent different kinds of functional genes. The guanine-cytosine content is indicated by darker grey and the adenine-thymine content is indicated by light grey.
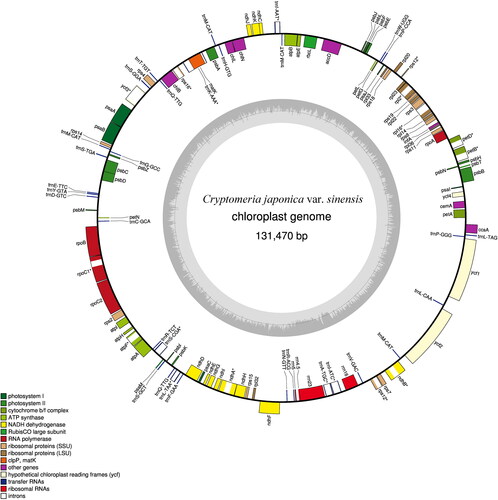
Table 3. Distribution of tRNA isotypes and anti-codons in the cp genomes of conifer species.
Chloroplast genome variation between wild and cultivated accessions
To investigate levels of cp sequence divergence between wild and cultivated trees, the nucleotide variation of 12 cp genomes was established. The results showed that wild and cultivated accessions possessed the same level of nucleotide variation (0.00003) (). We identified 11 and 10 mutation sites in the cp genome of wild and cultivated trees, respectively. Using the C. japonica cp genome (AP010967) as a reference, we identified a total of 29 or 28 InDels as well as 16 or 14 single-nucleotide polymorphisms (SNPs) (A/T) in wild and cultivated trees, respectively (). The trnL-ycf1 spacer had the highest number of indels (10), and the largest indel (198 bp) was found in ycf1, which is thought to be involved in cellular metabolism or to play a structural role in plastids [Citation36].
Table 4. Levels of nucleotide variation in the wild and cultivated C. japonica var. sinensis cp genomes.
Table 5. InDels in the wild and cultivated C. japonica var. sinensis cp genomes.
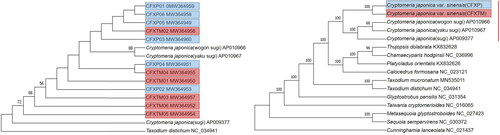
Intraspecific phylogeny analysis indicated that C. japonica var. sinensis and C. japonica trees did not fall into separate clades (). To determine the evolutionary relationship of C. japonica var. sinensis, we included 13 Cupressaceae species using 71 common protein-coding genes in the phylogenetic analysis. The result showed that wild and cultivated C. japonica var. sinensis trees formed a clade, which was sister to C. japonica with 100% bootstrap support. C. japonica var. sinensis and C. japonica had a close genetic relationship with Thujopsis dolabrata ().
Figure 2. Phylogenetic relationships based on protein-coding genes by maximum likelihood (ML) analyses. (A) Phylogenetic tree of the 12 C. japonica var. sinensis and C. japonica accessions. (B) Phylogenetic tree of the 13 Cupressaceae species.
Repeat sequences in chloroplast genome
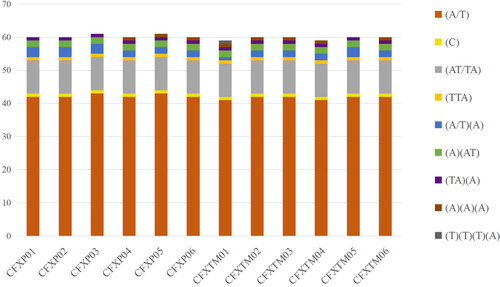
Chloroplast simple sequence repeats (cpSSRs) are used to investigate the levels of genetic diversity [Citation37–39]. In total, we detected 59-61 SSRs with a length ≥10 bp. Most of the repeated sequences were located in intergenic regions and only some in protein-coding sequences (, Supplemental file 2). This supports previous reports that SSR frequency varies between different regions of the genome [Citation40, Citation41]. Mononucleotide repeats were the most abundant SSRs, whereas no tetranucleotides were found (). Almost all SSRs were composed of A or T. These SSRs can function as useful molecular markers to explore population genetic structure and domestication events.
Figure 3. Number and distribution of simple sequence repeats (SSRs) in the 12 C. japonica var. sinensis cp genomes.
Conclusions
In this study, we reported 12 C. japonica var. sinensis cp genomes in wild and cultivated trees by de novo sequencing. The structure of the cp genome showed a partial lack of one IR copy, which is a common feature in gymnosperm cp genomes. Phylogenetic analysis suggested that the wild and cultivated trees possessed the same level of nucleotide variation. C. japonica var. sinensis had a close genetic relationship with T. dolabrata. Our study will be helpful for conserving this important timber forest species, and further studies.
Data accessibility
This work was supported by the National Key R&D Program of China (2016YFE0127200).
Acknowledgment
We are grateful to Dr. Markus Ruhsam and Dr. Berthold Heinze for checking English and valuable suggestions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Funding
All cp gemomes were uploaded to GenBank (accession no. MW364949-MW364960)
References
- Xue S, Shi T, Luo WJ, et al. Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Hortic Res. 2019;6(1):89.
- Huang H, Shi C, Liu Y, et al. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol Biol. 2014;14:151.
- Kuo LY, Tang TY, Li FW, et al. Organelle genome inheritance in Deparia ferns (Athyriaceae, Aspleniineae, Polypodiales). Front Plant Sci. 2018;9:486
- Droogenbroeck BV, Maertens I, Haegeman A, et al. Maternal inheritance of cytoplasmic organelles in intergeneric hybrids of Carica papaya L. and Vasconcellea spp. (Caricaceae Dumort., Brassicales). Euphytica. 2005;143(1-2):161–168.
- Ennos RA, Sinclair WT, Hu XS, et al. Using organelle markers to elucidate the history, ecology and evolution of plant populations. In Molecular systematics and plant evolution. London: Taylor & Francis; 1999. p. 1–19.
- Shinozaki K, Ohme M, Tanaka M, et al. The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J. 1986;5(9):2043–2049.
- Ohyama K, Fukuzawa H, Kohchi T, et al. Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature. 1986;322(6079):572–574.
- Li JJ, Zhang D, Ouyang KX, et al. The complete chloroplast genome of the miracle tree Neolamarckia cadamba and its comparison in Rubiaceae family. Biotechnol Biotechnol Equip. 2018;32(5):1087–1097.
- Xie WW, Li JN, Ye BJ, et al. The complete chloroplast genome of Cryptomeria japonica var. sinensis (Cupressaceae). Mitochondrial DNA B Resour. 2020;5(3):3392–3411.
- Wang WC, Chen SY, Guo W, et al. Tropical plants evolve faster than their temperate relatives: a case from the bamboos (Poaceae: Bambusoideae) based on chloroplast genome data. Biotechnol Biotechnol Equip. 2020;34(1):482–493.
- Kaila T, Chaduvla PK, Rawal HC, et al. Chloroplast genome sequence of clusterbean (Cyamopsis tetragonoloba L.): genome structure and comparative analysis. Genes. 2017;8(9):212.
- Xiao PG, Huang B, Ge GB, et al. Interspecific relationships and origins of Taxaceae and Cephalotaxaceae revealed by partitioned Bayesian analyses of chloroplast and nuclear DNA sequences. Plant Syst Evol. 2008;276(1-2):89–104.
- Sugiura M. The chloroplast genome. Plant Mol Biol. 1992;19(1):149–168.
- Sigmon BA, Adams RP, Mower JP. Complete chloroplast genome sequencing of vetiver grass (Chrysopogon zizanioides) identifies markers that distinguish the nonfertile’ Sunshine’ cultivar from other accessions. Ind Crops Prod. 2017;108:629–635.
- Strauss SH, Palmer JD, Howe GT, et al. Chloroplast genomes of two conifers lack a large inverted repeat and are extensively rearranged. Proc Natl Acad Sci USA. 1988;85(11):3898–3902.
- Jia XM, Liu CP. Characterization of the complete chloroplast genome of the Chinese yew Taxus chinensis (Taxaceae), an endangered and medicinally important tree species in China. Conservation Genet Resour. 2017;9(2):197–199.
- Yu T, Huang BH, Zhang YY, et al. Chloroplast genome of an extremely endangered conifer Thuja sutchuenensis Franch.: gene organization, comparative and phylogenetic analysis. Physiol Mol Biol Plants. 2020;26(3):409–418.
- Tsumura Y, Yoshimura K, Tomaru N, et al. Molecular phytogeny of conifers using RFLP analysis of PCR-amplified specific chloroplast genes. Theor Appl Genet. 1995;91(8):1222–1236.
- Patel RK, Jain M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS One. 2012;7(2):e30619.
- Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19(5):455–477.
- Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34(12):3299–3302.
- Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20(17):3252–3255.
- Lowe TM, Chan PP. tRNAscan-SE On-line: Search and Contextual Analysis of Transfer RNA Genes. Nucleic Acids Res. 2016;44(W1):W54–57.
- Lagesen K, Hallin P, Rødland EA, et al. RNammer: consistent annotation of rRNA genes in genomic sequences. Nucleic Acids Res. 2007;35(9):3100–3108.
- Lohse M, Drechsel O, Kahlau S, et al. OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013;41(Web Server issue):W575–W581.
- Kumar S, Stecher G, Li M, et al. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549.
- Beier S, Thiel T, Münch T, et al. MISA-web: a web server for microsatellite prediction. Bioinformatics. 2017;33(16):2583–2585.
- Hirao T, Watanabe A, Kurita M, et al. Complete nucleotide sequence of the Cryptomeria japonica D. Don. chloroplast genome and comparative chloroplast genomics: diversified genomic structure of coniferous species. BMC Plant Biol. 2008;8(1):70.
- Duan H, Guo JB, Xuan L, et al. Comparative chloroplast genomics of the genus Taxodium. BMC Genomics. 2020;21(1):114.
- Hao ZD, Cheng TL, Zheng RH, et al. The complete chloroplast genome sequence of a relict conifer Glyptostrobus pensilis: Comparative analysis and insights into dynamics of chloroplast genome rearrangement in Cupressophytes and Pinaceae. PloS One. 2016;11(8):e0161809.
- Xu J, Yang C, Liao BS, et al. Panax ginseng genome examination for ginsenoside biosynthesis. Gigascience. 2017;6(11):1–15.
- Goodenbour JM, Pan T. Diversity of tRNA genes in eukaryotes. Nucleic Acids Res. 2006;34(21):6137–6146.
- Jones TE, Ribas PL, Alexander RW. Evidence for late resolution of the aux codon box in evolution. J Biol Chem. 2013;288(27):19625–19632.
- Mohanta TK, Khan AL, Hashem A, et al. Genomic and evolutionary aspects of chloroplast tRNA in monocot plants. BMC Plant Biol. 2019;19(1):39.
- Hiratsuka J, Shimada H, Whittier R, et al. The complete sequence of the rice (Oryza sativa) chloroplast genome: intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol Gen Genet. 1989;217:185–194.
- Kikuchi S, Bedard J, Hirano M, et al. Uncovering the protein translocon at the chloroplast inner envelope membrane. Science. 2013;339(6119):571–574.
- Flannery ML, Mitchell FJ, Coyne S, et al. Plastid genome characterisation in Brassica and Brassicaceae using a new set of nine SSRs. Theor Appl Genet. 2006;113(7):1221–1231.
- Yang AH, Zhang JJ, Yao XH, et al. Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplification in L. chinense. Am J Bot. 2011;98:123–126.
- Jiao Y, Jia HM, Li XW, et al. Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myrica rubra). BMC Genom. 2012;13(1):201.
- Shimada H, Sugiura M. Pseudogenes and short repeated sequences in the rice chloroplast genome. Curr Genet. 1989;16(4):293–301.
- Cardle L, Ramsay L, Milbourne D, et al. Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics. 2000;156(2):847–854.