
Abstract
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-Cas9 mediated gene editing systems have been used in a variety of organisms, including plants. This system can efficiently edit target genes to generate mutants, especially multiplex mutants, which are invaluable genetic materials for gene functional studies. Although the use of traditional hybridization technology to construct multiplex mutants has been widely used in plant research, the complex screening and identification, long construction cycle and other problems have always troubled researchers. Therefore, in the current plant science research, the use of CRISPR-Cas9 mediated multiplex genome editing to construct multiplex mutants has become a mainstream technology. Here, we designed an improved plant CRISPR-Cas9 mediated multiplex genome vector system. The In-fusion cloning technology was selected to conveniently assemble multiple tandem sgRNA (Single-guide RNA) expression frames. In this system, Cas9-free plants can be easily selected by examining mCherry fluorescence in T2 transgenic plants. By using this system, Cas9-free homozygous triple mutant was obtained, demonstrating the availability of our system.
Introduction
The research on gene function in plant science directly affects the breeding of new crop varieties. At present, the study of gene function is inseparable from the use of mutants as materials. The functional redundancy and complex interaction between genes lead to the fact that a single-gene mutant often fails to satisfy the needs of plant science research and breeding. In the old days, the use of traditional hybridization technology to construct multiplex mutants was widely used in research, but researchers have been plagued by problems such as gene linkage, complicated screening and identification and long construction cycle. Therefore, using CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-Cas9 mediated gene editing to construct multiplex mutants has become a mainstream technique in current plant science research.
In the CRISPR-Cas9 system, sgRNA (Single-guide RNA) can guide Cas9 protein to specifically recognize and cut the target sequence of DNA to form a DSB (double-strand break). When the DSB is repaired by the organism itself, it is often accompanied by changes in the base sequence, which destroys the open reading frame of the original gene and leads to the functional loss of the target gene [Citation1]. SgRNA is a small length RNA that is less than 100 bp, so researchers can easily co-express multiple sgRNAs and Cas9 to achieve multiplex genome editing. In plant research, the expression frames of Cas9 and multiple sgRNAs are usually constructed in the T-DNA (Transfer DNA) of binary vectors, and then are delivered into plants through Agrobacterium-mediated transformation. The multiple target genes-edited homozygotes are obtained by genotyping the progeny of transgenic plants [Citation2–7].
In the current research, different researchers have adopted different strategies to construct multiple sgRNA expression frames into the binary vectors. For instance, in the study of Gao and Zhao (2016), two different sgRNA expression frames were cloned into the Pme I site and Mfe I site in a binary vector respectively [Citation2]. For another example [Citation7], designed and assembled two tandem repeat sequences, each containing three sgRNA expression frames, which were inserted into two sites (site 1: Hind III/Xma I; site2: EcoR I/Kpn I) in the binary vector respectively [Citation7]. Recently, it has been reported that the tandem repeat sequence containing 12 different sgRNA expression frames has been successfully constructed into the binary vector [Citation5]. It can be seen from the above examples that when the number of sgRNAs co-expressed with Cas9 is particularly large, researchers generally adopt the strategy of constructing tandemly repeated sgRNA expression frames.
In the currently reported studies, the construction of tandem repeat sgRNA expression frames is a cumbersome assembly process. Multiple sgRNA expression frames are assembled via the use of multiple restriction enzymes [Citation7]; or the use of pairs of isocaudomer enzymes, such as Spe I/Nhe I, BamH I/Bgl II, Xba I/Nhe I and Xho I/Sal I [Citation5, Citation8, Citation9], or the use of Golden Gate ligation or Gibson Assembly [Citation2, Citation4]. The above methods must first construct a single sgRNA expression frame before assembling the tandem repeat structure. Here, we design a simple tandem repeat assembly strategy. The 20-base sgRNA sequence as the homologous sequence was used for In-Fusion ligation. By using In-Fusion cloning technology, the PCR (Polymerase Chain Reaction) products can be easily assembled into tandem repeat sgRNA expression frames.
Moreover, in recent years, researchers have increasingly tended to remove heterologously expressed Cas9 and sgRNA after gene editing. This is because of the following reasons. Firstly, Cas9 and sgRNA would cause the risk of off-target editing [Citation10–12]. Secondly, this is not conducive to molecular complementation in later research, because the complemented wild-type genes will be edited again and will lose their function. Thirdly, it is difficult to distinguish whether the gene editing in transgenic offspring is inherited from the parents or newly edited in the current plants. In addition, in agricultural production, plants containing Cas9 and sgRNA are usually limited as transgenic crops. At present, the most common way to screen Cas9-free gene editing lines is to use fluorescent proteins as reporter genes, such as the mCherry system [Citation2, Citation13] and GFP system [Citation5]. Here, we choose the mCherry system, in which the reporter gene mCherry was constructed into the Arabidopsis pAt2S3 promoter and specifically expressed in seed coat [Citation2, Citation13]. By using this system, Cas9-free lines can be easily selected in the T2 generation.
Materials and methods
Plant materials and growth conditions
The Arabidopsis materials used in this study were wild-type Columbia-0 ecotype (N6673 - single seed line [ABRC, Arabidopsis Bilological Resource Center, https://abrc.osu.edu/]). Before being planted on the medium, mature Arabidopsis seeds were immersed in water at 4 °C for 2 days, sterilized with 0.1% HgCl2 for 10 min and then washed 5 times with sterilized H2O. All Sterilized seeds were grown in MS plates (4.43 g/L MURASHIGE & SKOOG (MS) BASAL MEDIUM w/VITAMINS [PhytoTechnology Laboratories™, https://phytotechlab.guidechem.com/ M519], 1% w/v sucrose [Solarbio, http://www.solarbio.com/ S8271], 1% w/v Agar powder [Solarbio A8190], pH5.75). Germination and plant growth took place at 22 °C under long-day conditions (80 μmol/m2·s white light, 16 h light/8 h dark cycle). Seven-day-old seedlings were transferred to the soil and grown at 22 °C under long-day conditions.
Plasmid construction
The sequences of sgRNA expression frames contained in the two tool vectors pUC57-Amp-U6TU6P and pUC57-Kan-CRP were constructed into two vectors by gene synthesis [GENEWIZ, https://www.genewiz.com.cn/]. We added the homologous sequence on both sides of the restriction site Mss I of binary vector pHDE-35S-Cas9-mCherry to pUC57-Kan-CRP. The primers CRP clon-F and CRP clon-R can pair with the homologous sequence to clone sequence of tandem sgRNA expression frames and insert them into the binary vector by In-Fusion cloning technology conveniently.
The sources of other materials and reagents used in plasmid construction are as follows: Binary vector pHDE-35S-Cas9-mCherry [Citation2] [Addgene, http://www.addgene.org/ Plasmid NO. #78931]; In-Fusion® HD Cloning Kit [Clontech, https://www.takarabio.com/ Code No. 639648]; PrimeSTAR® GXL DNA Polymerase [TaKaRa, https://www.takarabio.com/ Code No. R050A]; FastDigest EcoR I [Thermo ScientificTM, https://www.thermofisher.cn/cn/zh/home.html FD0275]; FastDigest Mss I [Thermo ScientificTM FD1344].
To construct vector pHDE-35S-Cas9-mCherry-IAA32-IAA34-TMK1, multiple tandem sgRNA expression frames were assembled and inserted into binary vector pHDE-35S-Cas9-mCherry as described above. All the primers used to construct the vector are listed in Supplemental Table S1.
Genetic transformation and identification of gene editing
Agrobacterium-mediated transformation of Arabidopsis with the binary vector was performed using the floral dipping method as previously described [Citation14]. DNA was extracted from transgenic plants by the CTAB method. Genomic regions surrounding the IAA32, IAA34 and TMK1 target sites were amplified by PCR using the primer pairs listed in Supplemental Table S2. Targeted gene mutations were detected by aligning the sequencing chromatograms of these PCR products with the wild-type controls. All the primers were purchased from AuGCT DNA-SYN Biotechnology Co., Ltd (http://www.augct.com/). All the sequencing in this study was provided by AuGCT DNA-SYN Biotechnology Co, Ltd, by using an Applied Biosystems® 3730/3730xl DNA Analyzer (Applied Biosystems, https://www.thermofisher.cn/) and Sanger method.
Results
Two tool vectors were designed to construct tandem sgRNA expression frames
To conveniently perform multiplex gene editing, two tool vectors pUC57-Amp-U6TU6P and pUC57-Kan-CRP were designed ( for plasmid map and sequence). Through using these two vectors, we can easily construct tandem sgRNA expression frames. As shown in , n pairs of primers containing sgRNA sequence in the 5′-end were designed for amplifying the sequence containing the U6 Promoter, U6 Terminator and Scaffold elements by PCR. After that, these amplified DNA fragments were assembled into tandem sgRNA expression frames. Finally, the tandem sgRNA expression frames can be amplified by CRP clon-F and CRP clon-R (Supplemental Table S1), and then assembled with the binary vector which was digested by Mss I (). The binary vector pHDE-35S-Cas9-mCherry was used in this study containing Cas9-NLS driven by the Cauliflower mosaic virus (CaMV) promoter 35S and mCherry driven by the Arabidopsis seed coat specific promoter (pAt2S3) (Plasmid map and sequence reference [Citation2]). By using this strategy, the number of sgRNA expression frames can be designed freely according to the number of target sites that would be edited to realize multiplex genome editing.
Figure 1. Schematic diagram of the construction of tandem sgRNA expression frames by using In-Fusion technology. The assembly of CRISPR-Cas9 expression vector can be accomplished in three steps. (1) Using our designed tool vectors pUC57-Amp-U6TU6P and pUC57-Kan-CRP as templates, primers containing sgRNA sequence at 5’-end are used for cloning DNA fragments containing U6 Promoter, U6 Terminator and Scaffold elements, at both ends of which there are sgRNA modules. (2) All the DNA fragments are assembled into tandem sgRNA expression frames by using In-Fusion technology (Clontech). According to experimental requirements, different numbers of tandem sgRNA expression frames can be assembled, which are contained in the pUC57-Kan vector backbone. (3) To clone the tandem sgRNA expression frames into the CRISPR-Cas9 expression vector pHDE-35S-Cas9-mCherry, the fragment containing sgRNAs is amplified by PCR with CRP clon-F and CRP clon-R (Supplemental Table S1), and then it can be inserted in the Mss I site of CRISPR-Cas9 expression vector by In-Fusion technology. In order to prevent the amplification of non-specific DNA fragments, when cloning the DNA fragment containing the pUC57-Kan vector backbone in step (1), the template plasmid pUC57-Kan-CRP should be linearized with EcoR I. Primers CRP test-F and CRP test-R are used for sequencing identification (Supplemental Table S1).
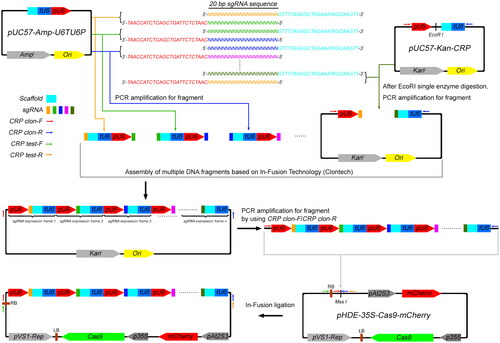
Figure 2. Workflow diagram of the transgene-free multiplex genome editing system. The construct with tandem multiple sgRNA expression frames was delivered into Arabidopsis through Agrobacterium-mediated transformation by floral dipping. The white arrows in the top panel show the seeds with red fluorescence in the T1 generation, and the white arrows in bottom panel show the seeds without red fluorescence in the T2 generation. (Scale bar, 1 mm.).
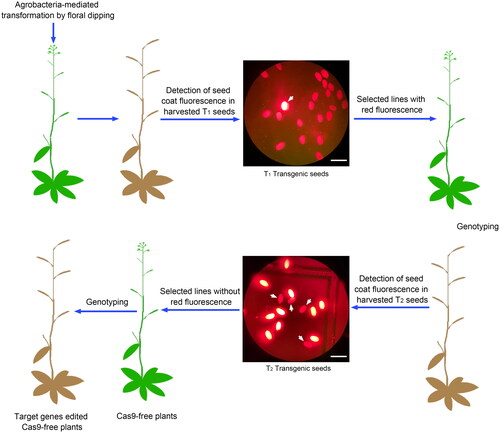
Workflow of selecting Cas9-free multiplex genome editing plants
In order to use seed coat reporter gene mCherry to generate Cas9-free plants with multiplex genome editing, the following workflow is used. Firstly, the binary vector inserted tandem multiple sgRNA expression frames is delivered into Arabidopsis by Agrobacteria-mediated transformation. Secondly, the harvested T1 transgenic seeds, which show red fluorescence, are selected by using a fluorescence microscope equipped with an mCherry filter (SUNNY CX40, Sunny Optical Technology(group) Co., Ltd, http://www.sunnyoptical.com/hd_362.html). When the selected T1 seeds grow up, the lines with gene editing at all target sites are selected by sequencing analysis, which are used to produce T2 generation seeds. Finally, in T2 transgenic plants, individuals without mCherry fluorescence are selected and genotyped to obtain multiplex genome editing mutants without exogenous Cas9 gene ().
Gene editing efficiency of three target sites was detected in T1 generation
To test the efficiency of our CRISPR-Cas9 system, IAA32 (INDOLE-3-ACETIC ACID INDUCIBLE 32), IAA34 and TMK1 (TRANSMEMBRANE KINASE 1) were chosen as targets for generating Cas9-free triple mutants. By using the vector construction scheme described above, the tandem sgRNA expression frames containing three sgRNAs were designed and inserted into the pHDE-35S-Cas9-mCherry binary vector (). Wild-type plants of Colombia ecotype were used for transformation. There were a total of 17 lines selected from the harvested T1 generation seeds, whose seed coat showed red fluorescence ().
Figure 3. Targeted gene modification of IAA32, IAA34 and TMK1 by using improved plant CRISPR-Cas9 vector system. (A) The schematics of the gene structure. CDS (Coding DNA Sequence) regions are shown in light blue and UTR (Untranslated Region) are depicted in red. Location and the sequences of the sgRNAs and PAM (Protospacer Adjacent Motif) sites are indicated. (B) The schematic of the CRISPR-Cas9 vector used to modify IAA32, IAA34 and TMK1. Arrows indicate the primers in this plasmid.
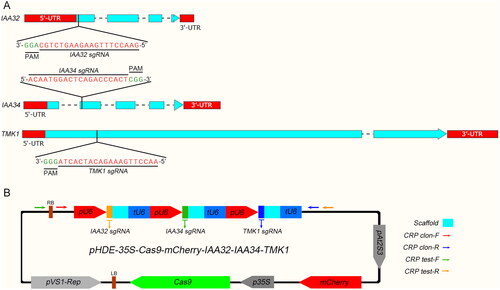
Figure 4. Results of selecting Cas9-free gene edited plants in T2 generation. The blue arrows represent the process of identification. (A) Statistical results of mCherry fluorescence in T1 generation and mutations at three loci in the lines with mCherry fluorescence. (B) Summary of the genotypes of T2 generation. The asterisk indicates that the homozygotes have two genotypes. (C) Sequencing results of three sgRNAs loci were obtained in T1 lines #4, #13 and #17 and T2 line #13.59.
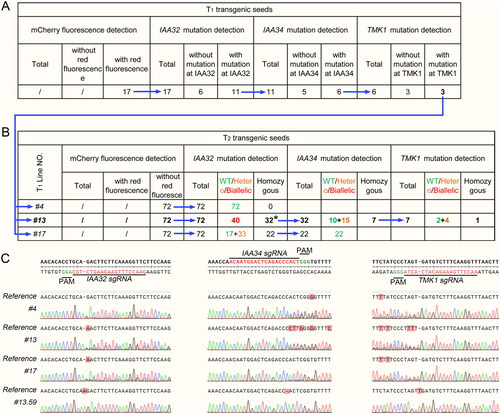
Then we identified the genotypes of these 17 lines. In the analysis of the results of Sanger sequencing, the overlap peak at the third or fourth base upstream of PAM (Protospacer Adjacent Motif) site was used as the basis to determine whether gene editing occurred. We conducted three rounds of identification of three gene editing targets. In each round of identification, the gene-edited lines detected were used as materials for the next round of identification. In the first round of identification of the IAA32 editing target, gene editing was detected in 11 out of 17 lines. In the second round of identification, the mutation of the IAA34 editing target was detected in 6 out of the 11 lines. In the last round of identification, gene editing of the TMK1 target site was detected in 3 out of the remaining 6 lines. These three lines with gene editing at all three target sites were numbered #4, #13 and #17, and the sequencing results near their sgRNAs and PAM sites are shown in .
Selection of Cas9-free triple mutants in T2 generation
In the identification of the transgenic T2 generation, we first used mCherry reporter gene to select seeds without exogenous gene Cas9 from three lines (). Similar to the identification of the T1 generation, we also conducted three rounds of identification. In each round of identification, individuals with homozygous mutation at the gene editing target sites were moved to the next round of identification.
Unfortunately, the results of line #4 in the first round of IAA32 genotyping and line #17 in the second round of IAA34 genotyping showed that they were all wild-type (). We speculated that the mutation caused by CRISPR-Cas9 mediated gene editing in these two T1 generation lines may be chimeric. Although gene editing was detected, these gene editing events occurring in somatic cells could not be passed on to the offspring. Through three rounds of identification, only line #13.59 with three homozygous mutation sites was isolated from the offspring of line #13 in T1 generation ().
Discussion
Gao et al. (2016) [Citation2], had used the binary vector pHDE-35S-Cas9-mCherry for Cas9-free gene editing, but this system is unsuitable for multiplex genome editing [Citation2]. Here, we designed a strategy, by using the ability of tandem sgRNA expression frames to be easily assembled and inserted into a binary vector for multiplex genome editing in Arabidopsis. Based on In-Fusion cloning technology, this strategy realizes homologous recombination of multiple DNA fragments and greatly simplifies the construction of tandem sgRNA expression frames. Theoretically, researchers can determine the number of tandem repeat sgRNA frames according to their own experimental needs by using our improved vector system.
From the example of using this system to construct multiplex mutants, we obtained a triple mutant with a single transformation experiment. In T1 generation, it was very easy to detect gene editing (sequencing results show overlapping peaks). The probability of gene editing detected in all three designed target sites was about 17.6% (3/17). However, in T2 generation, we screened only one triple mutant (#13.59) from the offsprings of three T1 lines (#4, #13 and #17), with a probability of about 0.5% (1/216) (). Obviously, the low probability is not conducive for researchers to use this system to construct more multiplex mutants than triple mutants. In view of this, some improved methods may be considered to use in the future research.
We already discussed the reasons why the genotypes identification of target sites in the T2 generation showed they were all wild type. Furthermore, in the first round for identifying the IAA32 target site of the T1 line #13 offspring, we detected homozygous mutations of two different genotypes, and the probability of homozygosity at this site was 44.4% (32/72). It is much higher than the probability of homozygous mutation identification at other sites. For example, the probability of homozygous mutation of T1 line #13 offspring at the IAA34 target site was 21.9% (7/32); in the offspring of T1 line #13 at the TMK1 target site it was 14.2% (1/7); in the offspring of T1 line #17 at the IAA32 target site it was 30.6% (22/72) (). We speculate that this is because both copies of IAA32 had been modified in the germ cells (megasporocytes and microsporocytes) of T1 line #13, and the forms of gene editing were different. This means that when two different homozygous mutations are isolated, other individuals have biallelic genotypes at the same time ().
According to the above analysis, the key of using this system to construct multiplex mutants is to increase the probability of heritable gene editing and bialleles by improving the efficiency of CRISPR-Cas9 mediated gene editing. First, a more efficient constitutive promoter can be used to replace the CaMV p35S to initiate the expression of Cas9, and the editing efficiency can be improved by increasing the abundance of Cas9 protein. For example, in the study of [Citation5], the UBQ10 promoter was used to initiate the expression of Cas9 [Citation5]. Secondly, sgRNA prediction tools can be used to select sites with high editing efficiency. Such as: http://crispr.hzau.edu.cn/cgi-bin/CRISPR2/CRISPR [Citation15]. Finally, multiple sgRNAs can be selected from each single gene for multiplex genome editing. By using this method, not only the editing efficiency can be improved, but also mutations with large fragments missing may be detected, which provides convenience for the identification of genotypes in the later stage [Citation16].
Conclusions
In our studies, an improved system was designed for CRISPR-Cas9 mediated multiplex genome editing in plant research. As mentioned above, the advantage of this system is that the tandem repeat sgRNA expression frames can be easily constructed. All fragments that assemble tandem repeats only need to be amplified by PCR. Researchers only need to add 20 base sgRNA sequences at the 5 ‘end of the primer of the fixed sequence to complete the primer design ( and Supplemental Table S1). In addition, mCherry was selected as the reporter gene in this system. With the help of a fluorescence microscope, genotyping can be effectively performed to select Cas9-free plants ().
Author contributions
L.W. conceived and directed the research; J.L.W., Y.L.L. and N.L. performed the major part of the research; Y.Y.L., W.G. and J.C. performed the experiments; Y.J.Z., H.Q.L. and Q.H.F. participated in the experimental discussion. L.W. wrote the draft of the paper and the final revision was accomplished by L.W., X.F.L. and G.Q.G. All authors have read and agreed to the published version of this manuscript.
Supplemental Material
Download Zip (781.1 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that supports the findings of this study are available in the supplemental data of this article.
Additional information
Funding
Related Research Data
References
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):1258096.
- Gao X, Chen J, Dai X, et al. An effective strategy for reliably isolating heritable and Cas9-Free arabidopsis mutants generated by CRISPR/Cas9-Mediated genome editing. Plant Physiol. 2016;171(3):1794–1800.
- Ma X, Liu YG. CRISPR/Cas9-Based multiplex genome editing in monocot and dicot plants. Curr Protoc Mol Biol. 2016;115:31 36 31–31 36 21.
- Ma X, Zhang Q, Zhu Q, et al. A robust CRISPR/Cas9 system for convenient, High-Efficiency multiplex genome editing in monocot and dicot plants. Mol Plant. 2015;8(8):1274–1284.
- Wang J, Chen H. A novel CRISPR/Cas9 system for efficiently generating Cas9-free multiplex mutants in arabidopsis. aBIOTECH. 2020;1(1):6–14.
- Xing HL, Dong L, Wang ZP, et al. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 2014;14:327.
- Zhang Z, Mao Y, Ha S, et al. A multiplex CRISPR/Cas9 platform for fast and efficient editing of multiple genes in arabidopsis. Plant Cell Rep. 2016;35(7):1519–1533.
- Wang C, Shen L, Fu Y, et al. A simple CRISPR/Cas9 system for multiplex genome editing in rice. J Genet Genomics. 2015;42(12):703–706.
- Yan L, Wei S, Wu Y, et al. High-Efficiency genome editing in arabidopsis using YAO promoter-driven CRISPR/Cas9 system. Mol Plant. 2015;8(12):1820–1823.
- Cho SW, Kim S, Kim Y, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24(1):132–141.
- Kuscu C, Arslan S, Singh R, et al. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol. 2014;32(7):677–683.
- Pattanayak V, Lin S, Guilinger JP, et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31(9):839–843.
- Yu H, Zhao Y. Fluorescence marker-assisted isolation of Cas9-Free and CRISPR-Edited arabidopsis plants. Methods Mol Biol. 2019;1917:147–154.
- Weigel D, Glazebrook J. In planta transformation of arabidopsis. CSH Protoc. 2006;2006(7):pdb.prot4668.
- Lei Y, Lu L, Liu HY, et al. CRISPR-P: a web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol Plant. 2014;7(9):1494–1496.
- Li JF, Norville JE, Aach J, et al. Multiplex and homologous recombination-mediated genome editing in arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol. 2013;31(8):688–691.