
Abstract
The combination drug menadione/ascorbate (M/A) manifests synergistic dose-dependent antiproliferative and cytotoxic effects towards cancer cells, but not towards normal cells of the same origin especially at concentrations that can be achieved in vivo by its oral and parenteral administration. It is assumed that M/A alters selectively dysfunctional cancerous mitochondria. However, the exact molecular mechanism is not clear yet. The aim of the present study was to elucidate the role of adenosine triphosphate (ATP) synthase activity and its suppression by oligomycin-A on M/A-induced cytotoxicity, mitochondrial superoxide and ATP level in leukaemic lymphocytes. Cells were treated with different concentrations of M/A in the absence and presence of oligomycin-A (100 ng/mL) for 24 h and 48 h. The cell growth and viability, steady-state ATP level and mitochondrial superoxide were analysed using conventional analytical tests. The results showed that suppression of ATP synthase activity by oligomycin-A decreased the cell growth and viability and increased the production of mitochondrial superoxide and depletion of ATP in cells treated with low/tolerable doses of M/A (up to 5/500 µM/µM), compared to the cells treated with M/A only. Oligomycin-A did not affect these parameters in cells treated with high doses of M/A (10/1000 and 20/2000 µM/µM). The inhibition of ATP synthase potentiated the cytotoxicity of M/A, particularly in leukaemic lymphocytes treated with low/tolerable doses. We assume that the cytotoxicity of M/A is tightly connected to impairment of oxidative phosphorylation, and mitochondrial ATP depletion is a crucial factor for cell death.
Introduction
Oxidative phosphorylation (OXPHOS) is the main source of energy (adenosine triphosphate (ATP)) in normal cells. Unlike them, most cancer cells are characterized by a unique shift in bioenergetics – reduction of OXPHOS and increased glycolysis, the so-called ‘Warburg effect’ [Citation1]. This compensatory switch is accompanied by some advantages: (i) maintenance of biosynthetic pathways during rapid cell proliferation (carbons from glucose and the Krebs cycle are used for amino acid, fatty acid and nucleic acid synthesis) and (ii) acidification of microenvironment due to increased lactate levels from glucose metabolism [Citation2]. Thus, via bioenergetics adaptation, cancer cells revive and sustain their survival and growth, which promotes their invasion and metastasis. Moreover, through metabolic ‘rearrangements’, cancer cells become more protected against conventional anticancer therapies. On the other hand, combined anticancer treatments, targeting both mitochondrial respiration and glycolysis, could be lethal to cancer cells [Citation3–5].
Until recently, it was generally accepted that cancer cells use glycolysis as a main source of ATP and do not rely on mitochondrial respiration for ATP production. However, recent studies demonstrated that the majority of ATP in cancer cells is produced by mitochondria, and some tumours show heavy dependence on OXPHOS [Citation6–8]. Therefore, blocking mitochondrial respiration should induce cell death in these tumours. These studies provide strong evidence that dysfunctional mitochondria are priority target to successful anticancer therapy. We believe that it is more correct to call cancerous mitochondria ‘altered’ instead of ‘dysfunctional’, because in fact they are functional.
Menadione (pro-vitamin K3) and ascorbate (vitamin C) interfere directly with the mitochondrial electron transport chain (ETC). Studies have demonstrated that naphthoquinones and benzoquinones affect mitochondrial respiration, bypassing Complex-I deficiency [Citation9–Citation11]. Pharmacological ascorbate and menadione are applied as adjuvants in the treatment of mitochondrial diseases [Citation11,Citation12]. The combination of ascorbate and menadione has been used clinically to bypass Complex-III deficiency of the ETC [Citation11,Citation12]. Both substances have been applied as a dietary supplement in combination with coenzyme Q10 (CoQ10), niacin, riboflavin and thiamin to bypass Complex-I and Complex-III of the ETC [Citation13]. Menadione is also a common supplement in animal feed (including diets for laboratory animals).
Menadione and ascorbate are redox-cyclers and administered alone, or in combination, they induce intracellular production of reactive oxygen species (ROS) by interaction with molecular oxygen [Citation14,Citation15]. The redox-active combination menadione/ascorbate (the ratio of 1/100 mol/mol menadione to ascorbate; M/A) has attracted the attention of researchers for more than 20 years due to its unusual ability to kill cancer cells without affecting the viability of normal cells, as well as with the synergistic anticancer effects of both molecules [Citation16–25]. This targeted cytotoxicity of M/A is accompanied by overproduction of mitochondrial superoxide and impairment of mitochondrial functionality in cancer cells only, assessed by a decrease in mitochondrial potential, NADH, NAD+ and oncometabolite succinate [Citation26,Citation27]. We assume that M/A-mediated suppression of cancer cell growth is a result of a specific mitochondrial redox-cycling between both substances, leading to severe oxidative stress and ATP depletion. We hypothesized that this specific redox-cycling of M/A occurs in the altered cancerous mitochondria only due to their overcharged and unbalanced ‘CoQ-pools’, overloading with metabolites (such as NADH and succinate), and ‘reversed’ Krebs cycle [Citation27,Citation28]. However, the exact molecular mechanism of M/A-mediated targeting and influencing OXPHOS in cancerous mitochondria is not clear yet.
Oligomycin-A is a mitochondrial ATP synthase inhibitor, widely used to suppress OXPHOS in living cells. It is considered that oligomycin-A blocks the proton channel, which significantly reduces the electron flow through the ETC, but it is not completely stopped due to a process known as ‘proton leak’ or ‘mitochondrial uncoupling’ – the protons return to the mitochondrial matrix independently of ATP synthase activity [Citation29]. In addition, a strong relationship between proton and electron leakage was demonstrated, which is explained by the sensitivity of superoxide production to reduced proton mobility due to its leakage [Citation30]. It was reported that treatment of cancer cells with oligomycin-A results in a decreased generation of ATP, which shifts their bioenergetics to glycolysis [Citation31]. We consider this as a suitable model to clarify whether the antiproliferative and cytotoxic effect of M/A is mediated by ATP synthase activity and OXPHOS.
The aim of the present study was to elucidate the role of ATP synthase activity and its suppression by oligomycin-A in M/A-induced cytotoxicity, mitochondrial superoxide and ATP level in leukaemic lymphocytes, as well as to clarify the role of mitochondrial ATP depletion in the antiproliferative and cytotoxic effects of this combination drug.
Materials and methods
Chemicals
Oligomycin-A was purchased from Selleck Chemicals (USA); menadione and ascorbate were purchased from Sigma-Aldrich (Weinheim, Germany). Other chemicals and kits were purchased from various suppliers. All reagents used in the experiments were analytical or HPLC grade.
Cells and treatment protocol
All experiments were performed on leukaemic lymphocytes derived from patients with acute lymphoblastic leukaemia (Jurkat; RIKEN Bioresource Center, Saitama, Japan). Cells were cultured in RPMI-1640 medium (Sigma-Aldrich, Weinheim, Germany), containing antibiotics (100 μg/mL of streptomycin and 100 U/mL of penicillin) (Sigma-Aldrich) and supplemented with 10% fetal bovine serum (FBS) (heat-inactivated) (Sigma-Aldrich). Leukaemic lymphocytes were grown in an incubator at 37 °C and a humidified atmosphere saturated with 5% CO2.
Before each experiment, the cells were sedimented by centrifugation (1000 × g/10 min) and placed in fresh medium without antibiotics. The lymphocytes were incubated with oligomycin-A (100 ng/mL) alone or in combination with menadione/ascorbate (M/A) for different time intervals, and at each time interval, aliquots were used for biochemical analyses. Ascorbate was dissolved in phosphate-buffered saline (PBS) (10 mM, pH 7.4). Oligomycin-A and menadione were dissolved in dimethyl sulfoxide (DMSO) (to 5 mM and 10 mM stock solutions, respectively) and then several working solutions in PBS were prepared. To avoid influence on cell viability, the final concentration of DMSO in the cell suspension was below 1%.
ATP assay
CellTiter-Glo™ Luminescent Cell Viability Assay (Promega, Madison, WI, USA) was used to analyse ATP levels in cell suspensions. The assay is based on the generation of a luminescent signal from a luciferin/luciferase reaction, which is proportional to the production of ATP in live cells [Citation32]. Shortly, aliquots of cell suspension (180 μL containing 1 × 106 cells/mL) were placed in 96-well transparent plates and incubated with 10 µL of oligomycin-A (final concentration 100 ng/mL) and 10 μL of M/A (at different concentrations − 2/200, 3/300, 5/500, 10/1000 and 20/2000 µM/µM) for 24 and 48 h, in a humidified atmosphere (at 37 °C, 5% CO2). A hundred microlitres of the cell suspension from each well were transferred to 96-well white plates, and 100 µL of the CellTiter-Glo reagent, containing luciferin and luciferase, were added to each well and incubated according to the manufacturer’s protocol. Luminescence was detected using a microplate reader (TECAN Infinite® M1000, Vienna, Austria), working in chemiluminescent mode.
Cell proliferation and viability assay
Cell viability and proliferation were analysed by trypan blue staining and automated cell counting, using Countess™ Automated Cell Counter (Invitrogen, Oregon, USA). Briefly, 10 μL of 0.4% trypan blue solution was added to 10 μL of cells suspension, incubated for 30 s, and 10 μL of cell suspension was placed in a Countess™ (Invitrogen) glass chamber. The number of live and dead cells was counted automatically. The linear range to operate with the automated cell counter was 1 × 104–5 × 106 cells/mL, and the optimal cell size was in the range of 5–60 μm.
Analysis of mitochondrial superoxide
A fluorogenic probe – MitoSOX™ Red Mitochondrial Superoxide Indicator (MitoSOX; Molecular Probes, Invitrogen, Eugene, Oregon, USA) was used for highly selective detection of superoxide in the mitochondria of live cells. The probe is cell-penetrating and localizes in the mitochondria. MitoSOX exhibits red fluorescence after oxidation by mitochondrial superoxide [Citation33].
Briefly, on the day of the experiment, MitoSOX™ Red was dissolved in DMSO to 5 mM stock solution, which was diluted with Hank’s Balanced Salt Solution (HBSS, containing Ca2+ and Mg2+) to prepare final 3 μM working solution. One-millilitre aliquots of cells (1 × 106 cells/mL) were collected via centrifugation, and the pellet was re-suspended in 1 mL of working MitoSOX solution. After 30 min at room temperature and protection from light, all samples were washed three times with PBS using centrifugation and finally re-suspended in 1 mL of PBS. The fluorescence intensity was detected immediately at λex = 510 nm and λem = 605 nm, using a microplate reader (TECAN Infinite® M1000, Vienna, Austria).
Statistical analysis
All data are expressed as mean values with standard deviation (±SD). Comparisons between the groups were performed using Student’s t-test. Differences were considered statistically significant at the level of p < 0.05.
Results and discussion
All analyses on M/A-treated leukaemic lymphocytes were carried out simultaneously in the absence and presence of oligomycin-A. Cell viability and proliferation was analysed by trypan blue staining and automated counting of live/dead cells in the samples. This analytical test allows differentiation of the cytotoxic from antiproliferative effect of the drug.
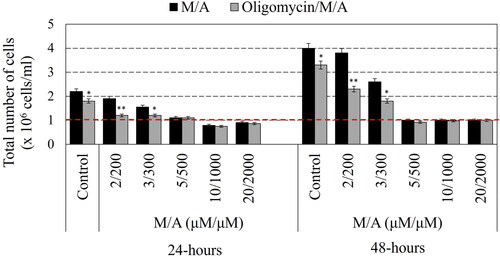
M/A (in concentrations from 2/200 to 20/2000 µM/µM) suppressed the proliferation of leukaemic lymphocytes at 24 and 48 h of incubation (). High doses of M/A (≥5/500 µM/µM) possessed a strong antiproliferative and even cytotoxic effect. The initial cell number in all suspensions was 1 × 106 cells/mL. In , the values below the red dashed line indicate a cytotoxic effect, while the values above the red dashed line indicate an antiproliferative effect (decreasing, but not stopping proliferation). Oligomycin-A (100 ng/mL) applied alone also suppressed the growth of leukaemic lymphocytes, but this effect was relatively slightly expressed. In the combination, oligomycin-A affected the effect of M/A on cell growth and viability in two ways: (i) it potentiated the antiproliferative effect of low/tolerable doses of M/A (<5/500 µM/µM); (ii) but it did not affect the antiproliferative and cytotoxic effect of high doses of M/A ( – grey columns versus black columns).
Figure 1. Effect of oligomycin-A (100 ng/mL) on the cell viability and proliferation of M/A-treated leukaemic lymphocytes (Jurkat) after 24 h and 48 h of incubation in humidified atmosphere. Data are means ± SD from three independent experiments with two parallel measurements for each experiment. *p < 0.05, **p < 0.01: oligomycin/M/A-treated versus the respective M/A-treated cells or oligomycin-treated sample versus the untreated cells (control). Red dashed line indicates the initial cell number in the suspension.
The effect of M/A on the steady-state level of ATP in the cancer cells also was dose-dependent and correlated with the number of cells in the respective sample and their viability ( and ). Data from ATP analysis were processed in two ways: (i) as absolute values of ATP-based luminescence in treated samples and untreated controls and (ii) as ATP-based luminescence normalized to the same number of living cells in each sample, since only living cells synthesize ATP. This approach for data processing was described in details in our previous study [Citation27]. M/A provoked a significant decrease in ATP-based luminescence in leukaemic lymphocytes, especially at high doses (≥5/500 µM/µM of M/A) (). Oligomycin-A (applied alone) also decreased the absolute steady-state level of ATP in the cells, but this effect was slightly expressed. In the combination, oligomycin-A further increased the ATP depletion in M/A-treated leukaemic lymphocytes, especially in the concentration range of 2/200–5/500 µM/µM. By normalizing the data to an equal number of living cells, two opposite trends were observed in M/A-treated cells. At high concentrations of M/A (≥10/1000 μM/μM), ATP-based luminescence decreased with time of incubation, which indicates a decrease in the number of living cells in the suspensions, suggesting cytotoxicity of M/A. The absolute values of ATP-based luminescence decreased in a concentration-dependent manner after 24 h of incubation. However, at 48 h of incubation with 2/200 and 3/300 μM/μM of M/A, the normalized ATP-based luminescence was slightly above the control level. In the case of 5/500 μM/μM of M/A-treated cells, the normalized value was below the control level, but slightly above the level detected at 24 h of incubation. At both time-intervals, oligomycin-A increased the ATP depletion in M/A-treated living cells, even at high doses of M/A. The data in could be interpreted in several aspects: (i) low/tolerable concentrations of M/A did not significantly affect the steady-state levels of ATP in leukaemic lymphocytes (in particular Jurkat), but the same concentrations decreased the proliferative activity, based on both assays; (ii) a concentration of 5/500 μM/μM of M/A is the critical concentration for the transition from antiproliferative to cytotoxic effect in the same cells and (iii) oligomycin-A further decreased the steady-state ATP levels in M/A-treated leukaemic lymphocytes, and this effect did not depend on the data processing mode.
Figure 2. Effect of oligomycin-A (100 ng/mL) on the absolute steady-state ATP levels in M/A-treated leukaemic lymphocytes (Jurkat) after 24 h and 48 h of incubation in humidified atmosphere. Data are means ± SD from three independent experiments with four parallel measurements for each experiment. *p < 0.05, **p < 0.01, p < 0.001: oligomycin/M/A-treated versus the respective M/A-treated cells or oligomycin-treated sample versus the untreated cells (control). ATP-based luminescence in the untreated cells was considered 100%.
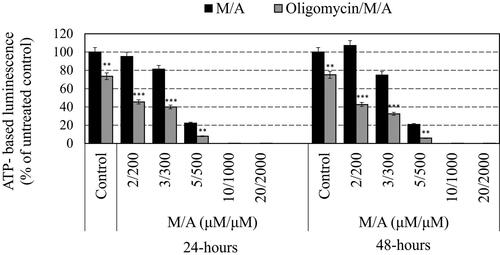
Figure 3. Effect of oligomycin-A (100 ng/mL) on the normalized steady-state ATP levels in M/A-treated leukaemic lymphocytes (Jurkat) after 24 h and 48 h of incubation in humidified atmosphere. Data are means ± SD from three independent experiments with four parallel measurements for each experiment. *p < 0.05, **p < 0.01, ***p < 0.001: oligomycin/M/A-treated versus the respective M/A-treated cells or oligomycin-treated sample versus the untreated cells (control). Data were normalized to an equal number of live cells in each sample (1 × 106 cells), as only living cells synthesize ATP. Normalized ATP-based luminescence in the untreated cells was considered 100%.
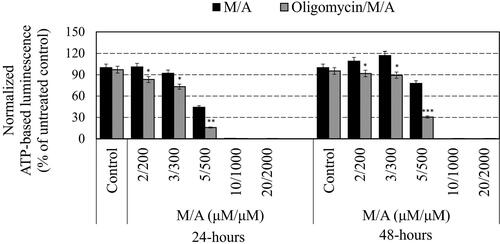
Studies have suggested that a menadione concentration of 5 µM is crucial for its mitochondrial redox-cycling [Citation11,Citation34,Citation35]. This concentration is considered a threshold level, since Complex-I bypass and ATP recovery in menadione-treated cells occurs only at concentrations below 5 µM [Citation11]. In our study, we observed that M/A at concentrations ≥5/500 µM/µM induces irreversible ATP depletion ( and – black columns), which could be a result of suppression of mitochondrial respiration due to irreversible ETC damage. However, at low/tolerable concentrations of M/A, the effect on ATP depletion and mitochondrial respiration is reversible. Inhibition of ATP synthase by oligomycin-A potentiates the M/A-mediated ATP depletion in leukaemic lymphocytes, which is evidence for mitochondrial mechanism of its antiproliferative and cytotoxic effects.
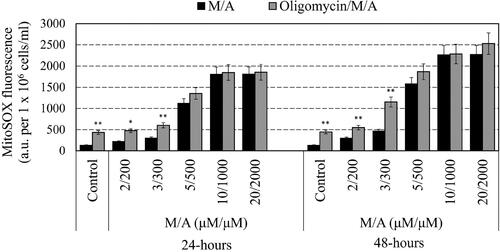
To investigate the effect of M/A and/or oligomycin-A on mitochondrial superoxide, we used fluorogenic probe MitoSOX Red Superoxide Indicator. The sensor easily crosses the cell membrane and localizes mainly in the mitochondria. In the absence of superoxide, MitoSOX has no fluorescence, but fluorescence appears after oxidation of MitoSOX by superoxide. The results are shown in . In M/A-treated leukaemic lymphocytes, the superoxide level increased in a dose-dependent and time-dependent manner: from ∼2 times for low/tolerable concentrations of M/A to ∼10 times for high concentrations of M/A within 48 h of incubation. Incubation of cells with oligomycin-A also induced production of mitochondrial superoxide (∼3 times over the control level). The combination oligomycin/M/A induced an additive increase in mitochondrial superoxide at M/A concentrations below 5/500 µM/µM. Oligomycin-A did not affect the level of mitochondrial superoxide in cells treated with high doses of M/A (≥5/500 µM/µM).
Figure 4. Effect of oligomycin-A (100 ng/mL) on the mitochondrial superoxide of M/A-treated leukaemic lymphocytes (Jurkat) after 24 h and 48 h of incubation in humidified atmosphere. Data are means ± SD from three independent experiments with two parallel measurements for each experiment. *p < 0.05, **p < 0.01: oligomycin/M/A-treated versus the respective M/A-treated cells or oligomycin-treated sample versus the untreated cells (control).
Our data suggest that inhibition of ATP synthase by oligomycin-A potentiated the antiproliferative and cytotoxic effect of M/A, particularly in leukaemic lymphocytes treated with low/tolerable doses, which was accompanied by ATP depletion and overproduction of mitochondrial superoxide. We assume that the antiproliferative and cytotoxic effects of M/A towards cancer cells are mediated by mitochondria-dependent mechanism(s), in particular by mitochondrial ATP synthesis.
It is interesting to note that a slight relapse was observed in cell proliferation and viability of M/A-treated cells in the absence and presence of oligomycin-A within the incubation time (48 h versus 24 h of incubation) (). This observation raises the question of whether leukaemic lymphocytes can reorganize and adapt their bioenergetics after M/A-treatment, shifting from OXPHOS to other pathways for ATP synthesis, when ATP synthase activity is inhibited by oligomycin-A.
Hao et al. [Citation31] have demonstrated that cancer cell lines with higher respiration rate show larger gains in glycolytic flux upon oligomycin-A treatment (100 ng/mL), which is an indicator for the compensatory role of glycolysis in energy supply [Citation31]. Inhibition of ATP synthase by oligomycin-A was also found to trigger fatty acid oxidation (FAO) as another alternative mechanism for ATP synthesis in cancer cells [Citation36]. Based on these observations, we assume that proliferation recovery after M/A-treatment of leukaemic lymphocytes could be due to activation of regulatory mechanism(s) related to reorganization and adaptation of their bioenergetics to glycolysis and/or FAO after oligomycin-A application. The increased generation of mitochondrial superoxide in oligomycin/M/A-treated cells compared to M/A-treated, especially at low/tolerable doses of M/A, is a factor disrupting the ETC, leading to additional decrease in proliferation activity and cell survival.
The findings from this study support our assumption [Citation27] that the possible reasons for the specific overproduction of mitochondrial superoxide in M/A-treated cancer cells are as follows: (i) a direct impairment of mitochondrial ETC by compromising its functionality, mainly Complex-I and Complex-III that are known to produce superoxide [Citation37,Citation38] and (ii) a specific mitochondrial redox-cycling of both substances, mediated by dysfunctional (altered) mitochondria, but not by the mitochondria of non-transformed cells [Citation11,Citation27]. Our hypothesis is that cancerous mitochondria could be discharged and offloaded by redox-cycling between menadione and ascorbate in them [Citation27]. Once superoxide exceeds a certain threshold, vital functional mitochondrial units (such as ETC, ATP synthase and others) can be destroyed, which causes inhibition of mitochondrial ATP synthesis, release of caspase-independent apoptosis-inducing factor (AIF) and cell death most likely via autoschizis [Citation39–41]. Inhibition of ATP synthase by oligomycin-A may exacerbate these effects, especially at low/tolerable M/A concentrations, potentiating the impairment of mitochondrial functionality in cancer cells.
Conclusions
Suppression of ATP synthase activity by oligomycin-A decreased the proliferation and viability and increased the production of mitochondrial superoxide and depletion of ATP in leukaemic lymphocytes treated with low/tolerable concentrations of M/A (up to 5/500 µM/µM), compared to the cells treated with M/A only. We assume that the cytotoxicity of M/A towards cancer cells is tightly connected to alteration of vital functional mitochondrial units (such as ETC, ATP synthase and others), which is accompanied by impairment of OXPHOS, and mitochondrial ATP depletion is one of the crucial factors for cell death.
Authors’ contributions
SS, ZZ and RB produced the first draft of manuscript. SS, DI, BN, GZ, RB and ZZ conducted the experiments. IA was involved in the critical review of the drafts and final version. All authors read and approved the final version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data that support the findings of this study are available on request from the corresponding authors (ZZ and BN).
Funding
This study was partially supported by the IC-MedTech Co. (USA) (ICM/QST grant, granted to R.B.) and the Japanese Agency for Medical Research and Development (AMED) (Project for Cancer Research and Therapeutic Evolution, P-CREATE, No. 16 cm0106202h0001).
References
- Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314.
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, et al. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20.
- Palorini R, Simonetto T, Cirulli C, et al. Mitochondrial complex I inhibitors and forced oxidative phosphorylation synergize in inducing cancer cell death. Int J Cell Biol. 2013;2013:243876:
- Liu H, Hu YP, Savaraj N, et al. Hypersensitization of tumor cells to glycolytic inhibitors. Biochemistry. 2001;40(18):5542–5547.
- Fath MA, Diers AR, Aykin-Burns N, et al. Mitochondrial electron transport chain blockers enhance 2-deoxy-d-glucose induced oxidative stress and cell killing in human colon carcinoma cells. Cancer Biol Ther. 2009;8(13):1228–1236.
- Fan J, Kamphorst JJ, Mathew R, et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. 2013;9:712.
- Vazquez F, Lim J-H, Chim H, et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013;23(3):287–301.
- Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11(1):9–15.
- Vafai SB, Mevers E, Higgins KW, et al. Natural product screening reveals naphthoquinone complex I bypass factors. PLoS One. 2016;11(9):e0162686.
- Majamaa K, Rusanen H, Remes A, et al. Metabolic interventions against complex I deficiency in MELAS syndrome. Mol Cell Biochem. 1997;174(1–2):291–296.
- Chan TS, Teng S, Wilson JX, et al . Coenzyme Q cytoprotective mechanisms for mitochondrial complex I cytopathies involves NAD(P)H: quinone oxidoreductase 1(NQO1)). Free Radic Res. 2002;36(4):421–427.
- Eleff S, Kennaway NG, Buist NR, et al. 31P NMR study of improvement in oxidative phosphorylation by vitamins K3 and C in a patient with a defect in electron transport at complex III in skeletal muscle. Proc Natl Acad Sci U S A. 1984;81(11):3529–3533.
- U.S. Department of Health and Human Services (FDA) Home Page; National Institutes of Health (Office of Dietary supplements). Dietary Supplements for Primary Mitochondrial Disorders. Available from: https://ods.od.nih.gov/factsheets/PrimaryMitochondrialDisorders-HealthProfessional/ (accessed on 10 March 2021).
- Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta. 2012;1826(2):443–457.
- Aljuhani N, Michail K, Karapetyan Z, et al. The effect of bicarbonate on menadione-induced redox cycling and cytotoxicity: potential involvement of the carbonate radical. Can J Physiol Pharmacol. 2013;91(10):783–790.
- Calderon PB, Cadrobbi J, Marques C, et al. Potential therapeutic application of the association of vitamins C and K3 in cancer treatment. Curr Med Chem. 2002;9(24):2271–2285.
- Marriage B, Clandinin MT, Glerum DM. Nutritional cofactor treatment in mitochondrial disorders. J Am Diet Assoc. 2003;103(8):1029–1038.
- Calderon PB, Verrax J, Taper HS. Targeting cancer cells by an oxidant-based therapy. Curr Mol Pharmacol. 2008;1(1):80–92.
- Taper HS. Altered deoxyribonuclease activity in cancer and its role in non-toxic adjuvant cancer therapy with mixed vitamins C and K3. Anticancer Res. 2008;28:2727–2732.
- Verrax J, Pedrosa RC, Beck R, et al. In situ modulation of oxidative stress: a novel and efficient strategy to kill cancer cells. Curr Med Chem. 2009;16(15):1821–1830.
- Calderon PB, Beck R, Glorieux C. Targeting Hsp90 family members: a strategy to improve cancer cell death. Biochem Pharmacol. 2019;164:177–187.
- Tareen B, Summers JL, Jamison JM, et al. A 12 week, open label, phase I/IIa study using apatone for the treatment of prostate cancer patients who have failed standard therapy. Int J Med Sci. 2008;5:62–67.
- Lasalvia-Prisco E, Cucchi S, Vazquez J, et al. Serum markers variation consistent with autoschizis induced by ascorbic acid-menadione in patients with prostate cancer. Med Oncol. 2003;20(1):45–52.
- U.S. National Library of Medicine Home Page. Double-blinded clinical trial using Apatone® for symptomatic postoperative total joint replacement (Apatone-B); ClinicalTrials.gov. Identifier:NCT01272830; Available from: https://clinicaltrials.gov/ct2/show/NCT01272830?term=apatone&draw=2&rank=1 (accessed on 10 March 2021).
- Noto V, Taper HS, Jiang YH, et al. Effects of sodium ascorbate (vitamin C) and 2-methyl-1,4-naphoquionone (vitamin K3) treatment on human tumor cell growth in vitro. I. Synergism of combined vitamin C and K3 action. Cancer. 1989;63(5):901–906.
- Semkova S, Zhelev Z, Miller T, et al. Menadione/ascorbate induces overproduction of mitochondrial superoxide and impairs mitochondrial function in cancer: comparative study on cancer and normal cells of the same origin. Anticancer Res. 2020;40(4):1963–1972.
- Bakalova R, Semkova S, Ivanova S, et al. Selective targeting of cancerous mitochondria and suppression of tumor growth using redox-active treatment adjuvant. Oxid Med Cell Longev. 2020;2020:6212935.
- Bakalova R, Zhelev Z, Miller T, et al. New potential biomarker for stratification of patients for pharmacological vitamin C in adjuvant settings of cancer therapy. Redox Biol. 2020;28:101357.
- Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, et al. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67.
- Papa S, Skulachev VP. Reactive oxygen species, mitochondria, apoptosis and aging. Mol Cell Biochem. 1997;174(1–2):305–319.
- Hao W, Chang CP, Tsao CC, et al. Oligomycin-induced bioenergetic adaptation in cancer cells with heterogeneous bioenergetic organization. J Biol Chem. 2010;285(17):12647–12654.
- Crouch SP, Kozlowski R, Slater KJ, et al. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J Immunol Methods. 1993;160(1):81–88.
- Dikalov SI, Harrison DG. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid Redox Signal. 2014;20(2):372–382.
- Levraut J, Iwase H, Shao ZH, et al. Cell death during ischemia: relationship to mitochondrial depolarization and ROS generation. Am J Physiol Heart Circ Physiol. 2003;284(2):H549–H558.
- Teixeira J, Amorim R, Santos K, et al. Disruption of mitochondrial function as mechanism for anti-cancer activity of a novel mitochondriotropic menadione derivative. Toxicology. 2018;393:123–139.
- De Oliveira MR, Liesa M. The role of mitochondrial fat oxidation in cancer cell proliferation and survival. Cells. 2020;9(12):2600.
- Dubouchaud H, Walter L, Rigoulet M, et al . Mitochondrial NADH redox potential impacts the reactive oxygen species production of reverse electron transfer through complex I. J Bioenerg Biomembr. 2018;50(5):367–377.
- Robb EL, Hall AR, Prime TA, et al. Control of mitochondrial superoxide production by reverse electron transport at complex I. J Biol Chem. 2018;293(25):9869–9879.
- Aguilar-Quesada R, Munoz-Games JA, Martin-Oliva D, et al. Modulation of transcription by PARP-1: consequences in carcinogenesis and inflammation. Curr Med Chem. 2007;14(11):1179–1187.
- Porter AG, Urbano AG. Does apoptosis-inducing factor (AIF) have both life and death functions in cells? Bioessays. 2006;28(8):834–843.
- Gilloteaux J, Jamison JM, Neal DR, et al. Cell damage and death by autoschizis in human bladder (RT4) carcinoma cells resulting from treatment with ascorbate and menadione. Ultrastruct Pathol. 2010;34(3):140–160.