
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Coxsackieviruses are distributed worldwide and can cause serious diseases threatening human health and even human life. Genetic silencing is considered a new opportunity to treat viral infections. In this study, we produced in vitro dsRNAs and siRNAs specific for two sequences of different length from the VP1 genetic regions of CVB3, and introduced them into HEp-2 cells with Oligofectamine as the transfection agent. We used the neurotropic strain Nancy. The 100% inhibitory concentrations of the applied dsRNAs and siRNAs were 0.3 μmol/L and fell in the range of non-cytotoxic concentration values. The produced VP1-specific dsRNAs and siRNAs were effective at low concentrations, suggesting that the produced RNAs are safe to use and possess excellent potential to treat CVB3 infection.
Introduction
Coxsackieviruses are distributed worldwide, although there are significant geographical differences with various serotypes. These differences are caused by climate types, public hygiene, the general public resistance and other factors [Citation1, Citation2]. It is considered that a great part of the diseases caused by the Coxsackievirus infections results from the tissue-specific collapse of the cells analogical to the cytopathic changes in cell cultures. The clinical manifestations of Coxsackievirus infection can lead to various disease syndromes. The primary infection usually occurs in the respiratory tract or the gastrointestinal epithelium, followed by viremia, which can eventually lead to a secondary infection [Citation3]. Secondary spread of the virus in the central nervous system can lead to aseptic meningitis and rarely to encephalitis or paralysis. An infection in the cardiovascular tissues can lead to pleurodynia or myocarditis. A disseminated infection can lead to exanthema, non-specific myalgia and severe multiple organ disease [Citation5–8]. Serious outcomes occur in the cases of Coxsackievirus infection during pregnancy, such as spontaneous abortions, fetal myocarditis and neurodevelopmental delays in the newborns [Citation9, Citation10]. Infections in newborns can have a fatal outcome. In infants infected with Coxsackievirus, there is a higher risk of potentially fatal complications, such as myocarditis, meningitis and encephalitis. Many chronic diseases, such as chronic myocarditis [Citation11], encephalitis lethargica [Citation12] and amyotrophic lateral sclerosis [Citation13, Citation14], may be the consequence of a previous Coxsackievirus infection.
The Coxsackievirus B (CVB) group of viruses, and especially CVB3, commonly cause cardiac infections. These include chronic infections associated with a latent form of the virus, which can be induced by deletions in the 5′-untranslated region of the viral genome [Citation15]. Over the past decades, in vitro and in vivo studies have focused on the molecular mechanisms of the cardiac coxsackievirus pathogenesis. The investigations revealed that not only the cell lesions induced by the virus, but also the immune and the autoimmune mechanisms take part in the development of Coxsackievirus-induced myocarditis [Citation16, Citation17].
Genetic silencing is considered a new opportunity to treat viral infections. Successful applications of the technique have been reported for treatment of HIV-1, HBV, HCV, SARS-coronavirus, influenza virus and polioviruses [Citation18, Citation19]. Some of these studies have already reached the stage of a clinical trial. RNA genetic silencing is a new strategy for inhibiting CVB3 [Citation21–25, Citation56]. According to the initial reports, there is considerable antiviral activity when the siRNAs are specific for a coding region of the viral genome, but not for non-translated regions. Moreover, silencing should target the viral plus chain because the minus chain may be inaccessible to the genetic silence apparatus [Citation20]. The viral genome is translated into a long polyprotein which is a subject to a series of cleavages by the viral proteases. The resulting mature viral proteins (For review see Citation26] include VP1-VP4 capsid proteins and seven non-structural proteins [Citation27]. After translation of the viral protein, the negative-strand replication follows [Citation28]. A viral replication complex forms to produce both positive- and negative-strand synthesis at the 5′ cloverleaf structure of the 5′ NTR (non-translating region) [Citation29]. In this way, CVB maximises its replication by occupying nearly all of the available cell resources [Citation30].
In this study, we produced specific dsRNAs and siRNAs-pools targeting two sequences from the VP1 genetic region of CVB3 and induced gene silencing in vitro.
Materials and methods
Materials
Cell cultures
Monolayer cell cultures of human epithelial cells from a larynx carcinoma (HEp-2) served for the in vitro experiments. Monolayers were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 5% fetal bovine serum in 96-well plates in a CO2- incubator at 37 °C and 5% CO2.
Virus
The neurotropic strain Nancy of Coxsackievirus B3 from the collection of the Department of Virology, Stephan Angeloff Institute of Microbiology, Bulgarian Academy of Sciences, Sofia, Bulgaria, was used in the experiments. The infectious titre in HEp-2 cells was 104.5 cell culture infectious dose 50% (CCID50).
Reference compounds
dsRNAs and siRNAs of the S-segment of bacteriophage Phi6 with lengths of 2948 bp and 25 bp, respectively (obtained from the Laboratory of Molecular Virology, University of Helsinki, Finland), served as reference compounds.
Methods
We adopted the procedures used in our previous paper [Citation31].
RNA extraction
Total RNA was extracted by the use of silica particles [Citation32]. Sixty grams of silica particles (S5631, Sigma-Aldrich Inc., USA) were added to 500 mL ultraclean water. The supernatant was removed after 24 h and another 500 mL of ultraclean water were added. The supernatant was removed after 5 h and the sediment was adjusted to pH 2.0 with HCl. Aliquots of 200 mL were autoclaved at 121 °C for 20 min and stored at 4 °C until use.
A volume of 500 mL cell suspension was homogenised with 500 mL Grinding buffer (GB) containing 4.0 mol/L guanadine thiocyanate, 0.2 mol/L Na-acetate (pH 5.2), 25 mmol/L ethylenediaminetetraacetic acid (EDTA), 1 mmol/L K-acetate and 2.5% (w/v) polyvinylpyrrolidone (PVP 40). Then, 100 mL 10% (w/v) N-lauril sarcosyl was incubated with 500 mL of the mixture at 70 °C for 10 min on a shaker, left for 5 min in ice and centrifuged at 11 330 g for 10 min at room temperature (RT). Next, 300 mL of the supernatant was mixed with 150 mL ethanol, 300 mL of 6 mol/L NaJ (in 0.15 mol/L Na2SO3) and 25 mL silica particles and incubated at RT for 10 min on a shaker. After centrifuging at 2415 g for 1 min, the supernatant was removed and the silica sediment was resuspended in 500 mL Washing Buffer (WB) containing 10 mmol/L Tris-HCl (pH 7.5), 0.5 mmol/L EDTA, 5 mmol/L NaCl and 50% (v/v) ethanol. This step was repeated twice. The sediment was left to dry at RT, resuspended in 150 mL of ultraclean water and incubated at 70 °C for 4 min. After centrifuging at 11 330 g for 3 min at RT, 130 mL of the supernatant was collected and stored at −20 °C.
Synthesis of DNA template
The amplification of two specific conserved fragments from VP1 (VP1a and VP1b) from CVB3 genome was performed by Touch-down RT-PCR. The primers were designed using viral sequences of CVB3 from the National Center for Biotechnology Information (NCBI) database and the BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) tools for alignment (, primers 1–4). A volume of 3 µL virus RNA was mixed with 7 µL of each relevant primer in a final volume of 10 µL, denaturated at 95 °C for 5 min and incubated with a master mix containing: 5 µL 5 MMLV-buffer, 2 µL dNTPs (2 mmol/L), 0.5 µL M-MuLV reverse transcriptase (200 U/µL), 7.5 µL DNase and RNase free H2O at 42 °C for 60 min to obtain copy DNA.
Table 1. Primers used in touch-down RT-PCR and synthesis of dsRNA.
The master mix for the following PCR was in a final volume of 25 µL and contained: 6 µL cDNA, 2.75 µL 10 PCR buffer, 2.2 µL MgCl2 (25 mmol/L), 2.2 µL dNTPs (2 mmol/L), 1 µL of each primer (10 µmol/L), 1 µL proofreading Pfu DNA-polymerase (5 U/µL) and 8.85 µL H2O). PCR was held in a thermocycler Auto-Q Server (LKB Instruments Ltd., UK) under the following conditions: initial denaturation at 95 °C for 3 min; 5 cycles of denaturation at 92 °C for 30 s, annealing at 64 °C for 30 s and elongation at 72 °C for 90 s; 5 cycles of denaturation at 92 °C for 30 s, annealing at 62 °C for 30 s and elongation at 72 °C for 90 s; 10 cycles of denaturation at 92 °C for 30 s, annealing at 60 °C for 30 s and elongation at 72 °C for 90 s; 10 cycles of denaturation at 92 °C for 30 s, annealing at 55 °C for 30 s and elongation at 72 °C for 90 s; and a final elongation at 72 °C for 10 min. The amplified DNA fragments were separated in 2% agarose gel in Tris-Acetic acid-EDTA (TAE) buffer with ethidium bromide (0.2 µg/mL) at 100 V for 1 h. PCR products were visualised in a transilluminator GenoPlex (VWR) with λ = 315 nm.
Synthesis of dsRNA and siRNA
Each primer (, primers 5–8) was designed to include 28–42 nucleotides of the target gene-specific sequence (VP1) at the 3′-end and an RNA-polymerase promoter at the 5′-end so that the resulting dsRNA PCR product contained the target sequence flanked by the promoter sequences. dsRNA was synthesised by a combination of in vitro transcription and replication from the DNA template following the instructions of the Replicator™ RNAi Kit (Finnzymes, Thermo Fisher Sceintific Inc., USA). The recombinant endo-ribonuclease from Giardia intestinalis - Power Cut Dicer was used to cut dsRNA into a pool of overlapping sequences of siRNAs with a length of approximately 20–30 nucleotides.
Transfection of cells
HEp-2 cells were transfected with dsRNAs/siRNAs using Oligofectamine™ Transfection Reagent (Invitrogen™, Thermo Fisher Sceintific Inc., USA) after plating in 100 mL DMEM 24 h before transfection, so that the cells were 30-50% confluent at the time of transfection.
Stock solutions for each transfection sample were prepared by diluting 1 μL of a 20 μmol/L stock (аnd the corresponding different concentrations used) oligonucleotide in 16 μL of serum-reduced DMEM to a final volume of 17 μL. The Oligofectamine™ Reagent was diluted 0.4–0.8 μL in serum-reduced DMEM to a final volume of 3 μL, incubated at RT for 5–10 min, added to the diluted oligonucleotide and incubated at RT for 15–20 min to form complexes. The growth medium from the cells was removed and the cells were washed once with serum-reduced DMEM. Another 80 μL of serum-reduced DMEM and 20 μL of complexes were added to each sample and incubated in a CO2-incubator at 37 °C for 4 h. Then, another 50 μL of DMEM containing 3x more serum were added. Assays were conducted 48 h post-transfection.
Determination of cytotoxicity of the dsRNAs and the siRNA pools
Trials were performed in four replicates. HEp-2 cells were cultured and incubated under the conditions described above for 12 h until formation of a confluent monolayer with a density of 2 × 105 cells/mL. The growth medium was removed and the cells were transfected with Oligofectamine™ containing dsRNAs or siRNAs in final concentrations 0.01, 0.03, 0.05, 0.1, 0.15, 0.2, 0.3, 0.9, 2.7, 8, 24 and 72 µmol/L. Then, the transfection medium was removed, the cells were incubated at 37 °C in 5% CO2 for 60 h and washed with 150 mL/well neutral red (NR) in concentration 0.4% stock solution diluted 1:80 with DMEM. Cells were incubated for another 3 h at 37 °C and 5% CO2 to let the dye penetrate the viable cells. The cells were washed again with 150 mL/well of phosphate buffered saline (PBS) and then, 100 mL/well of extraction solution (containing 1% glacial acetic acid, 50% ethanol and 49% distilled water) were added. The plates were shaken for 10 min and the absorbance of the samples was read on an ELISA Reader (Organon GmbH, Germany) at a wavelength of 540 nm and a reference value of 620 nm. The viability of the cells was calculated as a percent from the control (untreated) cells, which were considered as 100% viable, according to the formula:
(1)
(1)
where OD is the optical density (absorption) of the live cells.
Determination of the antiviral effect of the dsRNAs and the siRNA pools
Assays were performed in four replicates. HEp-2 cells were cultured for 12 h until formation of a confluent monolayer with a density of 2 × 105 cells/mL. Then, the growth medium was removed and the cells were transfected with Oligofectamine™ containing dsRNAs or siRNAs in final concentrations of 0.01, 0.03, 0.05, 0.1, 0.15, 0.2 and 0.3 µmol/L (the concentration range was selected according to the results obtained from the pilot pre-experimental test concentrations). The transfection medium was aspirated, the cells were inoculated with a virus dilution corresponding to a multiplicity of infection (MOI) 0.004 and left to absorb for 1 h. The unabsorbed virus was removed and the plates were incubated with DMEM for 35 h. Then, DMEM was removed and the cells were washed with 150 mL/well NR (0.4% stock solution diluted with DMEM at a ratio 1:80). The plates were incubated for another 3 h to let the dye penetrate the viable cells. The cells were washed again with 150 mL/well of PBS (without Ca2+ and Mg2+), the PBS was decanted and treated drop-wise with 100 mL/well of extraction solution (1% glacial acetic acid, 50% ethanol and 49% distilled water). After incubation for 10 min and the absorbance of the samples was read on an ELISA Reader (Organon GmbH, Germany) at a wavelength of 540 nm and a reference value of 620 nm [19]. Controls were healthy control (non-inoculated cells), virus control (virus-inoculated untreated cells) and cytotoxicity control (non-inoculated treated cells) cultivated under the same conditions, as well as internal controls (referent compounds). The antiviral effect was calculated as a percent according to the formula:
(2)
(2)
where OD is the optical density (absorption) of the live cells.
IC50 was recovered directly from the graphics using the linear function between the two nearest values in Microsoft Excel 2021 version 16. The selectivity index (SI) was calculated as:
(3)
(3)
where CC50 is the half-maximal cytotoxic concentration and IC50 is the 50% inhibitory concentration.
Results
The DNA template that was synthesised by RT-PCR using primers 1 and 2 had a length of 1000 bp. It was used to produce specific dsRNAs complementary to a longer fragment of VP1 (1080 bp) by the use of primers 5 and 6. The DNA template that was synthesised by RT-PCR using primers 3 and 4 had a length of 800 bp and flanked and was used to produce specific dsRNAs complementary to a shorter fragment of VP1 (820 bp) by the use of primers 7 and 8. The produced specific dsRNAs were the source for generating pools of overlapping siRNAs by the use of Power Cut Dicer endo-ribonuclease. The resulting dsRNAs and siRNAs-pools were tested for cytotoxicity and antiviral activity.
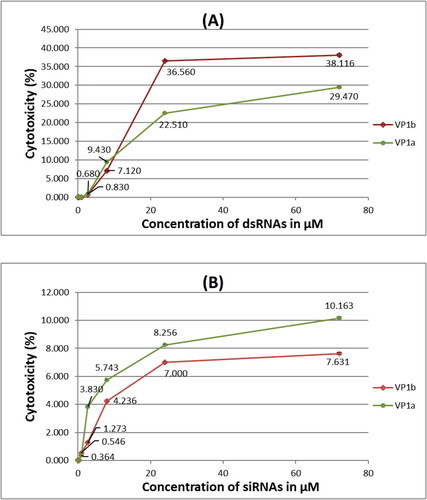
The highest concentration of dsRNAs/siRNAs tested for cytotoxicity (72 μmol/L) () exceeded more than 200-fold the concentration that gave 100% efficacy in inhibiting the virus infection. Cytotoxicity was not detected for concentrations up to 0.3 μmol/L (i.e. 100% cell viability) for all samples including dsRNAs and siRNAs specific for VP1 gene of the CVB3 (). The highest tested concentration of VP1-specific dsRNAs (72 μmol/L) gave below 40% cytotoxicity and the highest tested concentration of VP1-specific siRNAs (72 μmol/L) gave below 11% cytotoxicity. The tested concentrations of VP1a-specific dsRNAs were less cytotoxic compared to the dsRNAs specific for the longer region (VP1b) of the same gene at the higher concentrations. The tested concentrations of VP1b-specific siRNAs were less cytotoxic compared to the siRNAs specific for the shorter region (VP1a) of the same gene though not with a great difference. Generally, the VP1-specific siRNAs induced less damage to the cells than the specific dsRNAs with approximately 2.9 and 5 times for the longer and the shorter sequence, respectively, at the highest tested concentrations. Although the highest tested concentration was much higher than the concentration giving 100% efficacy in cell protection from the virus, we could not observe 50% cytotoxicity. At the same time, we were not able to produce more concentrated RNAs solutions, and consequently, it was not possible to calculate the CC50 value.
Figure 1. Cytotoxicity assay of different concentrations of dsRNAs (A) and siRNAs (B) specific for two sequences (a and b) of the VP1 genetic region of CVB3 in HEp-2 cells transfected with Oligofectamine™.
Note: Values are mean from four replicates.
The lowest concentration of dsRNAs targeting the VP1 gene with antiviral effect was 0.05 μmol/L, and 0.3 μmol/L fully inhibited CVB3 strain Nancy (). A concentration of 0.15 μmol/L VP1-specific dsRNAs inhibited the virus as much as 33% and 0.2 μmol/L − 75%. The linear trendline between these two nearest to 50% inhibition values returned the formula:
(3)
(3)
where y is the inhibition value (%) and x is the concentration of dsRNAs (μmol/L).
Figure 2. Antiviral assay of different concentrations of dsRNAs (A) and siRNAs (B) specific for two sequences (a and b) of the VP1 genetic region of CVB3 in HEp-2 cells transfected with Oligofectamine™.
Note: Values are mean from four replicates, rounded to an integer, with standard deviation error bars.
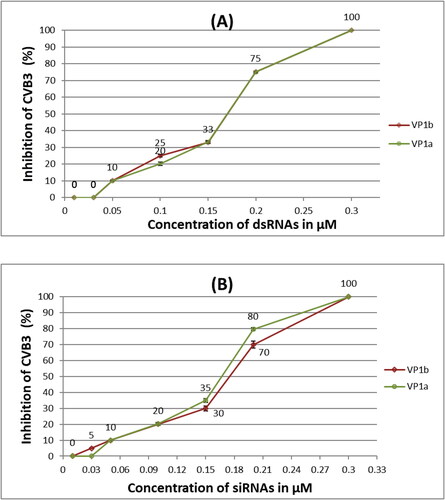
IC50 for the VP1-specific dsRNAs was calculated as 0.17 μmol/L.
The lowest tested concentrations (0.01 μmol/L) of the produced siRNAs also did not show any positive result in the inhibition of CVB3 Nancy (). A concentration of 0.05 μmol/L was able to protect 10% of the HEp-2 cells. Similarly to the tested dsRNAs, 0.15 μmol/L siRNAs inhibited the virus between 30-35% and 0.2 μmol/L − 70-80% (). The calculated (same as above) IC50 were expectedly with very close values − 0.167 μmol/L and 0.175 μmol/L for VP1a- and VP1b-specific siRNAs, respectively, derived from the functions:
(4)
(4)
(5)
(5)
where y is the inhibition value (%) and x is the concentration of siRNAs (μmol/L).
Overall, the 100% inhibitory concentrations of the applied dsRNAs and siRNAs targeting two sequences of the VP1 gene of CVB3 strain Nancy and using Oligofectamine as the transfection agent fell in the range of non-cytotoxic concentration values, which indicated the excellent potential of the produced RNAs to inhibit CVB3 infection. The selectivity indices (IS) were calculated as >424 (for dsRNAs), >431 (for VP1a-siRNAs) and >411 (for VP1b-siRNAs).
Discussion
To date, there is no approved antiviral therapeutic for treatment of CVB3 infection [Citation37]. RNA silencing of different regions of the virus have shown a potential for inhibition of virus infection and rational drug discovery [Citation21, Citation22, Citation24, Citation38, Citation39].
A major limitation in the long-term inhibition of viral infections is the emergence of resistant mutants which applies not only to the conventional low-molecular-weight antiviral therapy, but also to gene silencing. The accumulation of point mutations in or near the target region has been observed in various viruses [Citation40–42]. Three counter-strategies were proposed to overcome the viral resistance: (1) targeting conservative regions of the viral genome, (2) using a combination of effective antiviral siRNAs and (3) genetic attenuation of host cellular factors that are essential for the viral life cycle.
Suppression of host cell genes required by the virus to enter and replicate in cells gives a promising opportunity for sustaining inhibition of the viral spread. The advantage of this strategy is that viruses have a limited capacity to adapt to changes in host cells. The CAR receptor is essential for the entry of CVB3 in the cells and has been extensively investigated as a probable target [Citation24, Citation43–45]. However, CAR is strongly associated with the intercellular contacts of epithelial cells [Citation46, Citation47] and its prolonged attenuation can be dangerous. Asher et al. (2005) [Citation48] report embryonic death associated with heart defects in animals. At a later stage, CAR attenuation did not seem to induce obvious cardiac abnormalities [Citation49], but more detailed studies revealed disorders and blockage of atrioventricular conduction in adult mice [Citation50–52], as well as various phenotypic manifestations in other organs [Citation52]. Because the CAR receptor may have significant cellular function, its use as a therapeutic target is debatable [Citation53].
A systematic study of viral resistance in cell cultures has shown that viral inhibition with one or two different siRNAs fails through the emergence of resistant mutants. Only simultaneous administration of three siRNAs targeting different regions of the viral genome may maintain therapeutic efficacy. However, in the long run, mutated virus progeny still emerges though at very low levels [Citation54]. To overcome the limitation effects of mutations, the use of a cocktail (pool) siRNAs covering a 3.5 kb sequence of the CVB3 genome has been proposed [Citation55]. The pool was generated by synthesising two RNAs covering the region encoding most of the nonstructural CVB3 proteins, which were then cleaved to siRNAs by recombinant Dicer exoribonucleases. The use of a pool of siRNAs was significantly more effective than the use of single specific siRNAs, which was the reason to choose a pool of overlapping siRNAs of a selected genetic region in our studies.
Targeting coding regions as well as non-coding regions have been investigated. Merl and Wessely (2007) [Citation54] proposed that targeting genetic silence to coding regions of nonstructural proteins is a better choice than coding regions of structural proteins and that combined treatment with at least three different siRNA molecules overcomes loss of efficiency due to mutations. The 5′-non-translational region (5’UTR) of the CVB3 genome has also been targeted in different studies [Citation15, Citation23, Citation24, Citation54], however, this region contains internal ribosome attachment regions (IRES) which interfere with the attachment of the designed microRNAs (miRNAs) [Citation56] and significantly reduce the expected antiviral activity. Later experiments using a better selection of the sequence of 5’UTR of CVB3 revealed that this region still has a great potential to be used as a target for specific siRNAs [Citation39].
Studies on miRNAs targeting other viral regions have also shown their potential as targets for inhibition of viral replication [Citation24, Citation57, Citation58]. The cis-active replication element (CRE), located in the 2 C coding region of the CVB3 genome, is strictly conserved and siRNAs targeting this region provided a long-term protection against CVB3 viral infection [Citation59]. Application of siRNA targeting the 2 A protease gene led to a significant reduction in CVB3 infection in vitro [Citation38] and in vivo [Citation21]. Hydrodynamic transfection in mice reduced the viral myocarditis and the proinflammatory cytokines, and the survival rates increased from 20% to 50% ten days after infection [Citation25]. The findings of Racchi et al. (2009) [Citation60] confirmed that the protease 2 A is a particularly good target for CVB3. Petrov and Galabov (2016) [Citation39] achieved full inhibition of the virus in vitro using non-toxic concentrations of dsRNAs and siRNAs targeting the 2 A and the 3 D RNA-dependent RNA polymerase gene of CVB3. Other exploited regions are 3 C and the interspacer region between VP1 and VP3 genetic regions [Citation31, Citation39]. In this study, we targeted two sequences with different length from the VP1 gene of CVB3 and proved that this gene is also an excellent target for inducing genetic silencing.
Conclusions
The in vitro-produced dsRNAs and siRNAs, introduced in HEp-2 cells with Oligofectamine as the transfection agent, were able to fully inhibit the CVB3 strain Nancy infection. Concentrations of 0.3 μmol/L of dsRNAs and siRNAs specific for the VP1 gene of CVB3 provided full protection of the cells from the virus infection. The 100% inhibitory concentrations of the applied dsRNAs and siRNAs fell in the range of non-cytotoxic concentration values. The produced VP1-specific dsRNAs and siRNAs were effective at low concentrations, suggesting that the produced RNAs are safe to use and possess excellent potential to be explored further for the treatment of CVB3 infection.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study is available within the article.
Additional information
Funding
References (in the order of mention)
- Bergelson JM, Krithivas A, Celi L, et al. The murine CAR homolog is a receptor for coxsackie B viruses and adenoviruses. J Virol. 1998;72(1):415–419.
- Rossmann M, He Y, Kuhn R. Picornavirus-receptor interactions. Trends Microbiol. 2002;10(7):324–331.
- Oberste MS, Pallansch MA. Coxsackieviruses. In: Mahy BWJ, Van Regenmortel MHV, editors. Encyclopedia of virology. 3rd ed. Atlanta, GA: Academic Press; 2008. p. 580–587. https://doi.org/10.1016/B978-012374410-4.00372-1
- Semler BL, Ramsingh A. Coxsackievirus-induced pancreatitis. Viral Immunol. 2004;17(3):358–369.
- Huber S, Ramsingh A. Coxsackievirus-induced pancreatitis. Viral Immunol. 2004;17(3):358–369.
- Melnick J. Polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses. In: Fields BN, Knipe DM, Howley PM, editors. Fundamental virology. New York, NY: Raven Press; 1996. p. 655–712.
- Tebruegge M, Curtis N. Enterovirus infections in neonates. Semin Fetal Neonatal Med. 2009;14(4):222–227.
- Romero JR. Pediatric group B coxsackievirus infections. Curr Top Microbiol Immunol. 2008;323:223–239.
- Ornoy A, Tenenbaum A. Pregnancy outcome following infections by coxsackie, echo, measles, mumps, hepatitis, polio and encephalitis viruses. Reprod Toxicol. 2006;21(4):446–457.
- Euscher E, Davis J, Holzman I, et al. Coxsackie virus infection of the placenta associated with neurodevelopmental delays in the newborn. Obstet Gynecol. 2001;98(6):1019–1026.
- Chapman N, Kim K. Persistent coxsackievirus infection: Enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr Top Microbiol Immunol. 2008;323:275–292.
- Cree BC, Bernardini GL, Hays AP, et al. A fatal case of coxsackievirus B4 meningoencephalitis. Arch Neurol. 2003;60(1):107–112.
- Woodall CJ, Riding MH, Graham DI, et al. Sequences specific for enterovirus detected in spinal cord from patients with motor neurone disease. BMJ. 1994;308(6943):1541–1543.
- Woodall CJ, Graham DI. Evidence for neuronal localisation of enteroviral sequences in motor neurone disease/amyotrophic lateral sclerosis by in situ hybridization. Eur J Histochem. 2004;48(2):129–134.
- Kim SH, Jeong JH, Cho KC, et al. Target-specific gene silencing by siRNA plasmid DNA complexed with folate-modifi ed poly(ethylenimine). J. Control Release. 2005;104:223–232.
- McManus B, Chow L, Wilson J, et al. Direct myocardial injury by enterovirus: a Central role in the evolution of murine myocarditis. Clin Immunol Immunopathol. 1993;68(2):159–169.
- Huber S. Coxsackievirus-induced myocarditis is dependent on distinct immunopathogenic responses in different strains of mice. Lab Invest. 1997;76(5):691–701.
- Haasnoot J, Westerhout EM, Berkhout B. RNA interference against viruses: Strike and counterstrike. Nat Biotechnol. 2007;25(12):1435–1443.
- Mescalchin A, Restle T. Oligomeric nucleic acids as antivirals. Molecules. 2011;16(2):1271–1296.
- Schubert S, Rothe D, Werk D, et al. Strand-specific silencing of a picornavirus by RNA interference: evidence for the superiority of plus-strand specific siRNAs. Antiviral Res. 2007;73(3):197–205.
- Merl S, Michaelis C, Jaschke B, et al. Targeting 2A protease by RNA interference attenuates coxsackieviral cytopathogenicity and promotes survival in highly susceptible mice. Circulation. 2005;111(13):1583–1592.
- Ahn J, Jun ES, Lee HS, et al. A small interfering RNA targeting coxsackievirus B3 protects permissive HeLa cells from viral challenge. J Virol. 2005;79(13):8620–8624.
- Yuan J, Zhang J, Wong B, et al. Inhibition of glycogen synthase kinase 3beta suppresses coxsackievirus-induced cytopathic effect and apoptosis via stabilization of beta-catenin. Cell Death Differ. 2005a;12(8):1097–1106.
- Werk D, Schubert S, Lindig V, et al. Developing an effective RNA interference strategy against a plus-strand RNA virus: silencing of coxsackievirus B3 and its cognate coxsackievirus-adenovirus receptor. Biol Chem. 2005;386(9):857–863.
- Fechner H, Sipo I, Westermann D, et al. Cardiac-targeted RNA interference mediated by an AAV9 vector improves cardiac function in coxsackievirus B3 cardiomyopathy. J Mol Med. 2008;86(9):987–997.
- Sin J, Mangale V, Thienphrapa W, et al. Recent progress in understanding coxsackievirus replication, dissemination, and pathogenesis. Virology. 2015;484:288–304.
- Kitamura N, Semler BL, Rothberg PG, et al. Primary structure, gene organization and polypeptide expression of poliovirus RNA. Nature. 1981;291(5816):547–553.
- Gamarnik AV, Andino R. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 1998;12(15):2293–2304.
- Vogt DA, Andino R. An RNA element at the 5’-end of the poliovirus genome functions as a general promoter for RNA synthesis. PLoS Pathog. 2010;6(6):e1000936.
- Fujita K, Krishnakumar SS, Franco D, et al. Membrane topography of the hydrophobic anchor sequence of poliovirus 3A and 3AB proteins and the functional effect of 3A/3AB membrane association upon RNA replication. Biochemistry. 2007;46(17):5185–5199.
- Petrov N, Stoyanova M, Galabov A. Inhibition of coxsackievirus B3 cardiotropic strain woodruff replication by silencing essential viral genes. Biotechnol Biotechnol Equip. 2019;33(1):1582–1589.
- Rott ME, Jelkmann W. Characterization and detection of several filamentous viruses of cherry: adaptation of an alternative cloning method (DOP-PCR) and modification of an RNA extraction protocol. Eur J Plant Pathol. 2001;107(4):411–420.
- Woodruff JF. Viral myocarditis. A review. Am J Pathol. 1980;101(2):425–484.
- Fairweather D, Kaya Z, Shellam GR, et al. From infection to autoimmunity. J Autoimmun. 2001;16(3):175–186.
- Zhang XM, Li YC, Chen P, et al. MG-132 attenuates cardiac deterioration of viral myocarditis via AMPK pathway. Biomed Pharmacother. 2020;126:110091.
- Song JH, Ahn JH, Kim SR, et al. Manassantin B shows antiviral activity against coxsackievirus B3 infection by activation of the STING/TBK-1/IRF3 signalling pathway. Sci Rep. 2019;9(1):9413.
- Wei Y, Wang H, Xi C, et al. Antiviral effects of novel 2-Benzoxyl-Phenylpyridine derivatives. Molecules. 2020;25(6):1409.
- Yuan J, Cheung PK, Zhang HM, et al. Inhibition of coxsackievirus B3 replication by small interfering RNAs requires perfect sequence match in the Central region of the viral positive strand. J Virol. 2005b;79(4):2151–2159.
- Petrov N, Galabov A. Small interfering RNAs against human enteroviral infections. Acta Microbil Bulg. 2016;32(2):101–107.
- Gitlin L, Karelsky S, Andino R. Short interfering RNA confers intracellular antiviral immunity in human cells. Nature. 2002;418(6896):430–434.
- von Eije KJ, Ter Brake O, Berkhout B. Human immunodeficiency virus type 1 escape is restricted when conserved genome sequences are targeted by RNA interference. J Virol. 2008;82(6):2895–2903.
- Boden D, Pusch O, Lee F, et al. Human immunodeficiency virus type 1 escape from RNA interference. J Virol. 2003;77(21):11531–11535.
- Fechner H, Pinkert S, Wang X, et al. Coxsackievirus B3 and adenovirus infections of cardiac cells are efficiently inhibited by vector-mediated RNA interference targeting their common receptor. Gene Ther. 2007;14(12):960–971.
- Shi Y, Chen C, Lisewski U, et al. Cardiac deletion of the coxsackievirus-adenovirus receptor abolishes coxsackievirus B3 infection and prevents myocarditis in vivo. J Am Coll Cardiol. 2009;53(14):1219–1226.
- Sharma M, Mishra B, Saikia UN, et al. Inhibition of coxsackievirus infection in cardiomyocytes by small dsRNA targeting its cognate coxsackievirus adenovirus receptor. Indian J Med Res. 2017;146(4):520–527.
- Coyne CB, Bergelson JM. CAR: a virus receptor within the tight junction. Adv Drug Deliv Rev. 2005;57(6):869–882.
- Freimuth P, Philipson L, Carson SD. The coxsackievirus and adenovirus receptor. Curr Top Microbiol Immunol. 2008;323:67–87.
- Asher DR, Cerny AM, Weiler SR, et al. Coxsackievirus and adenovirus receptor is essential for cardiomyocyte development. Genesis. 2005;42(2):77–85.
- Chen JW, Zhou B, Yu QC, et al. Cardiomyocyte-specific deletion of the coxsackievirus and adenovirus receptor results in hyperplasia of the embryonic left ventricle and abnormalities of sinuatrial valves. Circ Res. 2006;98(7):923–930.
- Lisewski U, Shi Y, Wrackmeyer U, et al. The tight junction protein CAR regulates cardiac conduction and cell-cell communication. J Exp Med. 2008;205(10):2369–2379.
- Lim BK, Xiong D, Dorner A, et al. Coxsackievirus and adenovirus receptor (CAR) mediates atrioventricular-node function and connexin 45 localization in the murine heart. J Clin Invest. 2008;118(8):2758–2770.
- Pazirandeh A, Sultana T, Mirza M, et al. Multiple phenotypes in adult mice following inactivation of the Coxsackievirus and Adenovirus Receptor (Car) gene. PLoS One. 2011;6(6):e20203.
- Fechner H, Pinkert S, Geisler A, et al. Pharmacological and biological antiviral therapeutics for cardiac coxsackievirus infections. Molecules. 2011;16(10):8475–8503.
- Merl S, Wessely R. Anti-coxsackieviral efficacy of RNA interference is highly dependent on genomic target selection and emergence of escape mutants. Oligonucleotides. 2007;17(1):44–53.
- Nygardas M, Vuorinen T, Aalto AP, et al. Inhibition of coxsackievirus B3 and related enteroviruses by antiviral short interfering RNA pools produced using phi6 RNA-dependent RNA polymerase. J Gen Virol. 2009;90(Pt 10):2468–2473.
- Schubert S, Grunweller A, Erdmann VA, et al. Local RNA target structure influences siRNA efficacy: Systematic analysis of intentionally designed binding regions. J Mol Biol. 2005;348(4):883–893.
- Akashi H, Miyagishi M, Yokota T, et al. Escape from the interferon response associated with RNA interference using vectors that encode long modified hairpin-RNA. Mol Biosyst. 2005;1(5–6):382–390.
- Aigner A. Gene silencing through RNA interference (RNAi) in vivo: Strategies based on the direct application of siRNAs. J Biotechnol. 2006;124(1):12–25.
- Lee HS, Ahn J, Jee Y, et al. Universal and mutation-resistant anti-enteroviral activity: Potency of small interfering RNA complementary to the conserved cis-acting replication element within the enterovirus coding region. J Gen Virol. 2007;88(Pt 7):2003–2012.
- Racchi G, Klingel K, Kandolf R, et al. Targeting of protease 2A genome by single and multiple siRNAs as a strategy to impair CVB3 life cycle in permissive HeLa cells. Methods Find Exp Clin Pharmacol. 2009;31(2):63–70.