
Abstract
Neutrophil gelatinase-associated lipocalin (NGAL) is a component of the human innate immune system that exerts bacteriostatic effects. Recent research suggests that NGAL is also a promising biomarker for tubular damage and aids in the early diagnosis of acute kidney injury. Therefore, the development of a method to efficiently produce recombinant NGAL has considerable clinical value. This study aimed to analyze the correlation between fluorescence reporter (enhanced green fluorescent protein, EGFP) intensity and NGAL levels, mediated by a bicistronic expression system based on internal ribosome entry sites (IRESs). During the 14-day culture period, EGFP intensity was positively correlated with NGAL concentrations. The purified protein also exerted bacteriostatic effects, interfering with bacterial iron uptake machinery during periods of iron starvation. The purified NGAL was also applied to generate a standard curve that could be used for enzyme-linked immunosorbent assay of samples containing unknown NGAL quantities. In conclusion, we successfully developed a procedure using EGFP and IRES-based vector to predict NGAL production levels and optimize protein harvest time. Our approach is also a simple and effective method for evaluating bioactivity.
Introduction
Neutrophil gelatinase-associated lipocalin (NGAL), also known as lipocalin 2, is a secretory protein belonging to the functionally diverse lipocalin family. Its tertiary structure is an eight-stranded anti-parallel beta-barrel, with a binding site in the interior for small lipophilic ligands including retinoids, steroids, arachidonic acid and iron [Citation1,Citation2]. NGAL forms a complex with gelatinase-B or matrix metallopeptidase-9 (MMP-9), protecting MMP-9 from proteolytic degradation and increasing its enzymatic activity [Citation3]. Another one of NGAL’s functions is to sequester bacterial ferric siderophores and interfere with iron transport into microorganisms, thus inhibiting of bacterial growth [Citation4]. In patients with kidney disease, NGAL levels are associated with renal injury severity, especially acute kidney injury (AKI), reflecting the degree of tissue damage. This biomarker is measurable in peripheral blood and urine samples, in particular being easily detected in urine within 2 h following ischaemia [Citation5]. Investigation in 71 children undergoing cardiopulmonary bypass showed augmentation of plasma and urinary NGAL levels as early as 2 h after surgery in those who developed AKI. This increase in NGAL concentration precedes an increase in creatinine levels by 1–3 days after cardiopulmonary bypass [Citation6]. Increases in NGAL levels have also been observed in other clinical conditions, including septic AKI [Citation7], contrast-induced nephropathy [Citation8] and nephrotoxicity [Citation9]. As a result, NGAL is considered one of the most promising next-generation biomarkers in clinical nephrology [Citation10,Citation11].
Given its uses, recombinant NGAL production will greatly benefit the development of immunological approaches for AKI diagnosis, follow-up and prognosis. During recombinant production, fluorescent proteins are normally used as reporters fused with the target protein [Citation12]. Besides this fusion process, fluorescent proteins are also often modified with a peptide linker to maintain stability and functionality [Citation13]. In contrast, reporter genes can be separately expressed using individual promoters or an internal ribosome entry site (IRES) as a bicistronic vector [Citation14,Citation15]. When fused or co-expressed, the fluorescent reporter can be easily quantified and monitored along with the target protein, improving recombinant protein production. Accordingly, reporter expression should accurately reflect the expression of the gene of interest [Citation16,Citation17]. Through measuring reporter expression in an IRES system, we can determine the appropriate time for harvesting protein products and avoiding their degradation. This information also allows us to design strategies that optimize culture conditions [Citation18]. However, not all genes are equally useful in this system, with several studies reporting that genes downstream of IRESs have substantially attenuated expression compared with those upstream [Citation19,Citation20]. Additionally, enhanced green fluorescent protein (EGFP) activity in the second cistron of the recombinant protein was undetectable among cells transduced with IRES-based lentiviral vector [Citation21]. Therefore, it is necessary to test the expression of individual recombinant proteins.
Several methods for exploring the biological activity of recombinant NGAL have been established [Citation4,Citation22,Citation23]. A prominent one involves interference with siderophore-mediated iron uptake to inhibit bacterial growth [Citation4]. Under iron-deficient conditions, bacteria produce siderophores to scavenge iron from the extracellular milieu [Citation24]. NGAL can disrupt the siderophore system through sequestering the molecules and preventing their uptake by bacteria [Citation25,Citation26]. To assess NGAL interference of siderophores, the standard technique is to count colony forming units (CFUs) on viable plates [Citation22]. However, counting bacteria is not well suited for high-throughput screening because the procedure requires long incubation periods and large quantities of proteins. Instead, bacterial proliferation in recombinant NGAL systems could be more efficiently monitored with optical density (OD) measurements using spectrophotometry, already a reliable and routine procedure for evaluating bacterial cultures.
In this study, we examined the correlation between EGFP intensity and NGAL production using an IRES-based expression system. Upon the evaluation of protein bioactivity, the interference with Escherichia coli growth was determined in an iron-deficient medium and a protocol for the microtiter plate was also developed for an efficient expression process.
Materials and methods
Cell culture
Human embryonic kidney cells (HEK-293T) were cultured as a monolayer in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% foetal bovine serum (FBS), 2 mmol/L L-glutamine, and 100 U/mL penicillin/streptomycin. Cells were maintained at 37 °C in a humidified incubator containing 5% CO2.
Design of NGAL expressing bicistronic vector
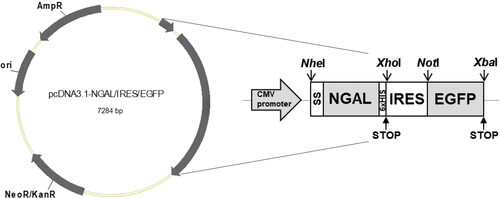
The complete coding sequence of human NGAL (NM_005564.4) was fused to a signal sequence (SS) at the 5’ end and linked with a 6×-His sequence, stop codon, IRES and EGFP sequences at the 3’ end (). Open reading frames were inserted into the NheI and XhoI cloning sites. This vector was then synthesized into a pcDNA3.1 vector by GenScript Biotechnology (Nanjing, China) and named pcDNA3.1-NGAL/IRES/EGFP.
Figure 1. Schematic diagram of the NGAL-expressing bicistronic vector. The tandem sequences (SS, human NGAL coding sequence, His6×, IRES and EGFP sequences) were inserted into pcDNA3.1 as the acceptor vector, with constitutive expression controlled via the cytomegalovirus (CMV) promoter.
Transient transfection in 293 T cells
Transient transfection was achieved using jetPRIME transfection reagent (Polyplus, New York, NY, USA), following the manufacturer’s protocol. Briefly, 5 × 106 293 T cells were plated onto a 10-cm culture dish containing DMEM supplemented with 10% FBS. For transfection, 10 µg of vector was mixed with 500 µL of buffer and 20 μL of jetPRIME™ transfection reagent and incubated at room temperature (RT) for 10 min. This mixture was added dropwise to 293 T cells in a serum-containing medium and distributed evenly, before gently agitating the plates and incubating them at 37 °C in 5% CO2. Two days after transfection, half of the culture supernatant was collected; fresh medium was added to the supernatant every 2 days, following a previously described procedure [Citation27]. Before replacing the medium, EGFP expression was observed under a fluorescence microscope (Optika Microscopes, Bergamo, Italy). Fluorescence intensity was quantified in ImageJ (http://rsb.info.nih.gov/ij/) through measuring the intensity of 20 individual cells per image and subtracting background fluorescence.
Quantitation of secreted NGAL using double-antibody sandwich ELISA
Secreted NGAL concentrations were determined using a Human Lipocalin-2/NGAL DuoSet ELISA kit (R&D Systems, Minneapolis, MN, USA, cat. no. DY1757-05). Briefly, 2 µg/mL of rat anti-human lipocalin-2 capture antibody diluted in phosphate buffered saline (PBS) was coated onto microtiter plates and left overnight at 4 °C in a moisture chamber. Coated wells were washed four times with washing buffer and incubated with blocking solution (1% bovine serum albumin [BSA] in PBS) at RT for 1 h. After washing, culture supernatants and recombinant NGAL standards were added to the wells and incubated at RT for 2 h. The plate was washed and further incubated with 25 ng/mL biotinylated goat anti-human lipocalin-2 detection antibody in a blocking solution at RT for 2 h. Next, the antibody-NGAL sandwich complex was detected using streptavidin conjugated to horseradish peroxidase (HRP). After a final wash, the TMB substrate was added. The reaction was stopped with the addition of 1 N HCl. OD at 450 nm was measured using a microplate reader (Bio-Rad, Hercules, CA, USA).
Protein purification using affinity chromatography
The supernatant was centrifuged at 300 ×g for 15 min to remove cell debris, then mixed with binding buffer at a 1:1 ratio and applied to a 5-mL nickel nitrilotriacetate (Ni-NTA, Ni-Sepharose) column equilibrated in 50 mmol/L sodium phosphate (pH 7.8). After extensive washing in the same buffer, elution was performed using a buffer containing 250 mmol/L imidazole (pH 7.4). The flow-through and elution fractions were analyzed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with Coomassie blue staining and western blotting. Fractions containing NGAL protein were concentrated via diafiltration using an Amicon Ultra 10 K centrifugal filter (Merck Millipore, Burlington, MA, USA).
SDS-PAGE and western blotting
SDS-PAGE was performed using 12% (w/v) separating and 4% (w/v) stacking gels. Next, proteins were transferred to PVDF membranes (GE Healthcare, Little Chalfont, UK), which were incubated with 2% BSA in TBS as a blocking buffer and washed with TBS containing 0.05% Tween 20. Reactive bands were detected using a mouse anti-His tag monoclonal antibody (ABclonal, Wuhan, China, cat. no. AE003) and probed with HRP-conjugated goat anti-mouse immunoglobulin G (Bio-Rad, Hercules, CA, USA, cat. no.1706516). Bands were visualized with a TMB membrane peroxidase substrate system (KPL, Gaithersburg, MD, USA).
Testing for bacteriostatic activity
A single fresh colony of E. coli (ATCC 25922) was precultured (37 °C, 200 rpm for 16 h) in polypropylene culture tubes containing the iron-limited minimal medium M9 (8.5 mmol/L NaCl, 31.1 mmol/L NH4Cl, 36.7 mmol/L KH2PO4, 70.4 mmol/L Na2HPO4, 0.5 mmol/L MgSO4, 0.15 mmol/L CaCl2 and 0.2% glucose). Cell densities of overnight cultures were initially adjusted to 107 CFU/well in a 96-culture plate, at 100 µL/well. Additional components up to 150 µL/well, 100 nmol/L NGAL and/or 0.1 µmol/L or 10 µmol/L FeCl3 were added to three independent wells for each combination or concentration tested. Plates were incubated again at 37 °C and 500 rpm for 12 h. Bacterial turbidity at OD595 was monitored at 6 and 12 h using a microplate reader (Bio-Rad, Hercules, CA, USA). The experiment was performed in triplicate and results were reported as fold-change relative to untreated conditions.
Setting up an ELISA standard curve
Purified NGAL was used to generate a standard curve for enzyme-linked immunosorbent assay (ELISA) to determine the NGAL concentration in spiked samples. Recombinant protein was serially diluted in blocking reagent (1% BSA in PBS) at concentrations ranging from 156.25 to 5,000 pg/mL. The ELISA procedure was performed as described above. Standard curves were prepared by plotting mean absorbance (from triplicate measurements) against the log of purified NGAL concentrations. Accuracy assessments involved analyzing spiked samples (200, 500 and 1,000 pg/mL diluted with the same matrix) derived from commercial recombinant protein and in the range of the standard curve. Measured concentrations were obtained from calibration curves. Percentage recovery was determined as follows: (measured concentration/spiked concentration) × 100.
Statistical analysis
The relationship between NGAL concentration and EGFP intensity was analyzed using Spearman’s correlation test. Bacteriostatic activity was compared using one-way analysis of variance (ANOVA), followed by Fisher’s LSD post hoc tests. Expected NGAL concentrations and concentrations in spiked samples were compared with the chi-square test. Statistical significance was set at p < 0.05.
Results
Correlation of EGFP intensity and NGAL concentration
Fluorescence intensity analysis revealed that EGFP expression was detected from post-transfection days 2 to 14 (). Fluorescence intensity was highest on day 4. This signal showed a declining trend (). Maximum NGAL production in the culture supernatant also occurred on day 4 and diminished with decreasing EGFP intensity.
Figure 2. Monitoring EGFP and NGAL expression. (A) Cell morphology and fluorescent protein expression. Transfected 293 T cells were observed under a fluorescence microscope for 14 days after transfection. Images were captured every 2 days using a 20× objective lens (scale bar: 50 µm). D, day. (B) Mean fluorescence intensity of EGFP and NGAL concentration. (C) Correlation between fluorescence intensity and NGAL concentration, calculated using Spearman’s rank correlation test. Results presented are from triplicate experiments (mean ± SEM).
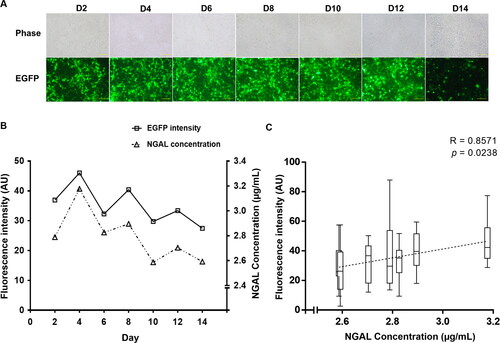
Statistical analysis confirmed that the correlation between EGFP intensity and NGAL concentration was direct (Spearman’s rho = 0.85, p < 0.05) ().
Purification of NGAL protein
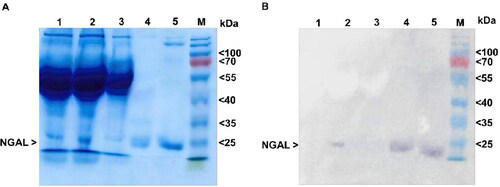
The results of SDS-PAGE revealed that the primary band in all eluted fractions was approximately 25 kDa (). Western blotting using a monoclonal anti-His-tag antibody confirmed the presence of this protein (). Reactive bands were found in the eluted fractions and pre-purified supernatant, with no protein degradation. After dialyzing the purified fraction using diafiltration, we measured NGAL to yield a final concentration of 1.6 µmol/L (5 µg protein yield per 10 cm tissue culture plate). These results demonstrate that recombinant NGAL was successfully purified with no degradation.
Figure 3. Identification of NGAL protein. (A) SDS-PAGE. Each sample was analyzed under reducing conditions. Lane 1: culture medium control, lane 2: pre-purified supernatant, lane 3: flow-through fraction, lane 4: first eluted fraction, lane 5: second eluted fraction, M: protein marker (kDa) (Thermo Fisher Scientific, Vilnius, Lithuania, cat. no. 26616). (B) Western blotting. The membrane was probed using an anti-His monoclonal antibody.
Iron sequestration of NGAL interferes with bacterial growth
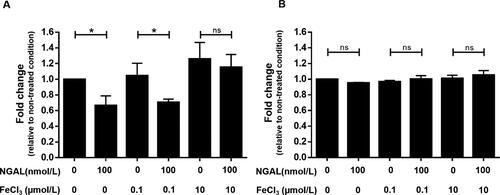
The results of testing bacteriostatic effects under iron-limiting conditions demonstrated that 100 nmol/L NGAL significantly inhibited bacterial growth at 6 h after inoculation (). The inhibitory effect was obvious at ferric chloride concentrations of 0 and 0.1 µmol/L, but not at 10 µmol/L, indicating that the bacteriostatic effect was iron-dependent. However, all inhibitory activity ceased at 12 h of culture (), reflecting a limited amount of NGAL.
Figure 4. Inhibition of bacterial growth in iron-limited culture. The effect of NGAL on bacterial growth at (A) 6 h and (B) 12 h after treatment. The growth of E. coli culture was defined by measuring turbidity at OD595. Each condition is normalized to OD595 for the non-treated condition (i.e. absence of NGAL and ferric chloride). Results are from triplicate experiments (mean ± SEM). Data were analyzed using one-way ANOVA, followed by post hoc LSD tests. *p < 0.05 and ns = not significant.
Purified NGAL used as calibrator in immunoassays
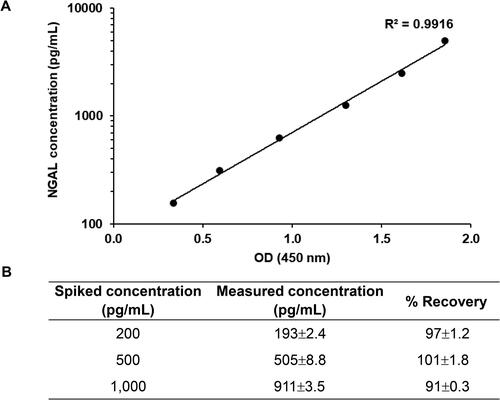
Purified NGAL was used as a calibrator in ELISA (). OD was correlated with the log of calibrator concentrations ranging from 156.25 to 5,000 pg/mL (R2 = 0.9916). Results from the spiked samples showed that measured NGAL levels did not differ significantly (p = 0.407) from spiked concentrations. Moreover, recovery percentage was 91–101% (). This result demonstrated that purified NGAL is suitable as a calibrator when using ELISA to measure NGAL concentrations in unknown samples.
Figure 5. Calibration curve constructed with purified NGAL. (A) Calibration curve for NGAL assay. Purified NGAL was serially diluted twofold and subjected to sandwich ELISA. All values represent the average of three replicates. Background OD values were subtracted from individual OD values. (B) Spiked and standard NGAL recovery concentrations. Three levels of spiked samples were analyzed in the same procedure as the standard protein. Recoveries for spiked samples compared to the measured recovery. Results presented are from triplicate experiments (mean ± SD).
Discussion
In this study, we aimed at the real-time monitoring of the production strategies for recombinant NGAL in mammalian culture by analyzing the expression of an EGFP reporter. It was explored whether the fluorescence intensity was related to the NGAL protein secretion levels. In this work, NGAL and EGFP were designed to be expressed under the same promotor in extracellular and cytoplasmic forms, respectively. Theoretically, a fusion protein, as a single chain, allows for a direct correlation between the fluorescence intensity of EGFP and the concentration of the protein of interest. In that format, however, the GFP fusion protein could affect the function of the target protein, its cellular processing and localization [Citation28]. Therefore, the bicistronic expression system mediated by the IRES sequence was applied to express these two proteins with a single promoter.
An alternative to bicistronic vectors that we considered was self-cleaving 2 A peptides. These molecules have some advantages because of their small size and stronger correlation with gene expression than IRES [Citation29]. Additionally, a study that monitored real-time fluorescent imaging found that gene expression dynamics are significantly more correlated for the multicistronic porcine teschovirus-1 2 A peptide-containing vector than the IRES-based vector, but only in HEK293 cells and not in Neuro2a cells. Thus, IRES activity seems dependent on cell type, whereas 2 A peptides are not [Citation30]. However, the 2 A-based cassette has a major disadvantage because it occasionally causes incomplete product cleavage, leading to decreased protein yield and protein dysfunction. Hence, we ultimately chose to use the IRES-based system.
As shown in the results, the intensity of the reporter protein showed a decreasing trend during the culture period. However, an analysis of the fluorescence intensity demonstrated that it was positively correlated with the amount of secreted NGAL throughout the production time. Our data imply that the expression of EGFP as a second cistron would be a good indicator for predicting protein production and also for optimizing the duration for protein harvest. According to this analytical approach, the optimal day for collection was 4 days after transfection (). With this strategy, assessing the fluorescence intensity proved to be much simpler, 10 times faster and with a higher reproductivity than the chloramphenicol acetyltransferase (CAT) assay [Citation31].
Bacteriostatic activity testing was used as a method to evaluate the biological function of the produced NGAL in terms of the inhibition of bacterial growth under iron-deprivation conditions. The underlying mechanism of this biological function is probably NGAL inhibition of siderophore-mediated iron uptake systems [Citation11]. However, a previous study showed that structural modifications in certain siderophores prevented NGAL binding [Citation32]. For example, NGAL was not recognized in gram-negative bacteria expressing iroA, including E. coli, Salmonella spp. and Klebsiella pneumoniae. Thus, the siderophore type should be considered when evaluating NGAL bioactivity.
In this work, the protocol for testing the inhibition of bacterial growth was adapted using a microplate together with the estimated cell density by measuring the turbidity in terms of OD instead of colony counting. This technique showed many benefits, including (i) providing a rapid and convenient method for investigating cell growth, since it only requires a microplate reader and (ii) culturing in 96-well plates leads to the utilization of only a small amount of protein at nanomolar concentrations. However, the use of the microplate photometer for reading the OD was limited by the range of cell numbers; therefore, titration was considered.
Accurately and efficiently determining NGAL levels has clinical importance, given the protein’s application as an early biomarker of AKI [Citation6]. Almost all available studies used ELISA to quantify NGAL [Citation33], meaning that a standard curve for this protein is needed to improve measurement accuracy. Here, we applied purified NGAL as a calibrator for ELISA and we found a good correlation between its concentration and OD. The high accuracy of measured spiked sample concentrations resulted in a reliable calibration curve. Moreover, the lowest concentration of this standard curve is on the level of picogram per millilitre, sensitive enough for detecting protein concentrations in the nanogram per milliliter among healthy subjects [Citation34].
Conclusions
In light of our present findings, it is conceivable that real-time measurements of fluorescence intensity are a good indicator of NGAL expression in an IRES-based system. These results provide empirical evidence to support the viability of a strategy for optimizing cell culture duration during protein harvesting. The simplicity of measuring EGFP provides an opportunity to apply flow sorting to positively select a single NGAL-overexpressing clone based on fluorescence intensity. Selection of high-producing clone based on flow cytometry and cell sorting has significantly increased the throughput of selection which can be screened in a shorter duration, leading to improved productivity.
Acknowledgment
The authors would like to thank Asst. Prof. Dr. Sarayot Rareongjai (Division of Clinical Immunology and Transfusion Science, Department of Medical Technology, School of Allied Health Sciences, University of Phayao, Phayao, Thailand) for his assistance with statistical analyses.
Disclosure statement
The authors declare that there are no conflicts of interest.
Data availability statement
All data that support this study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Kjeldsen L. Gelatinase granules in human neutrophils. Eur J Haematol Suppl. 1995;56:1–30.
- Schenkels LC, Veerman EC, Nieuw Amerongen AV. EP-GP and the lipocalin VEGh, two different human salivary 20-kDa proteins. J Dent Res. 1995;74(9):1543–1550.
- Yan L, Borregaard N, Kjeldsen L, et al. The high molecular weight urinary matrix metalloproteinase (MMP) activity is a complex of gelatinase B/MMP-9 and neutrophil gelatinase-associated lipocalin (NGAL). Modulation of MMP-9 activity by NGAL. J Biol Chem. 2001;276(40):37258–37265.
- Goetz DH, Holmes MA, Borregaard N, et al. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10(5):1033–1043.
- Mishra J, Ma Q, Prada A, et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol. 2003;14(10):2534–2543.
- Mishra J, Dent C, Tarabishi R, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet. 2005;365(9466):1231–1238.
- Bagshaw SM, Bennett M, Haase M, et al. Plasma and urine neutrophil gelatinase-associated lipocalin in septic versus non-septic acute kidney injury in critical illness. Intensive Care Med. 2010;36(3):452–461.
- Hirsch R, Dent C, Pfriem H, et al. NGAL is an early predictive biomarker of contrast-induced nephropathy in children. Pediatr Nephrol. 2007;22(12):2089–2095.
- McWilliam SJ, Antoine DJ, Jorgensen AL, et al. Urinary biomarkers of aminoglycoside-induced nephrotoxicity in cystic fibrosis: kidney injury molecule-1 and neutrophil gelatinase-associated lipocalin. Sci Rep. 2018;8(1):5094.
- Bolignano D, Donato V, Coppolino G, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a marker of kidney damage. Am J Kidney Dis. 2008;52(3):595–605.
- Schmidt-Ott KM, Mori K, Li JY, et al. Dual action of neutrophil gelatinase-associated lipocalin. J Am Soc Nephrol. 2007;18(2):407–413.
- Su WW. Fluorescent proteins as tools to aid protein production. Microb Cell Fact. 2005;4(1):12.
- Arai R, Ueda H, Kitayama A, et al. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001;14(8):529–532.
- Martinez-Salas E. Internal ribosome entry site biology and its use in expression vectors. Curr Opin Biotechnol. 1999;10:458–464.
- Bayat H, Hoseinzadeh S, Pourmaleki E, et al. Evaluation of different vector design strategies for the expression of recombinant monoclonal antibody in CHO cells. Prep Biochem Biotechnol. 2018;48(2):160–164.
- Liu S, Bugos RC, Dharmasiri N, et al. Green fluorescent protein as a secretory reporter and a tool for process optimization in transgenic plant cell cultures. J Biotechnol. 2001;87(1):1–16.
- March JC, Rao G, Bentley WE. Biotechnological applications of green fluorescent protein. Appl Microbiol Biotechnol. 2003;62(4):303–315.
- Cha HJ, Dalal NG, Pham MQ, et al. Insect larval expression process is optimized by generating fusions with green fluorescent protein. Biotechnol Bioeng. 1999;65(3):316–324.
- Zhou Y, Aran J, Gottesman MM, et al. Co-expression of human adenosine deaminase and multidrug resistance using a bicistronic retroviral vector. Hum Gene Ther. 1998;9(3):287–293.
- Mizuguchi H, Xu Z, Ishii-Watabe A, et al. IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol Ther. 2000;1(4):376–382.
- Yu X, Zhan X, D’Costa J, et al. Lentiviral vectors with two independent internal promoters transfer high-level expression of multiple transgenes to human hematopoietic stem-progenitor cells. Mol Ther. 2003;7(6):827–838.
- Miethke M, Skerra A. Neutrophil gelatinase-associated lipocalin expresses antimicrobial activity by interfering with L-norepinephrine-mediated bacterial iron acquisition. Antimicrob Agents Chemother. 2010;54(4):1580–1589.
- Gupta K, Shukla M, Cowland JB, et al. Neutrophil gelatinase-associated lipocalin is expressed in osteoarthritis and forms a complex with matrix metalloproteinase 9. Arthritis Rheum. 2007;56(10):3326–3335.
- Miethke M, Marahiel MA. Siderophore-based iron acquisition and pathogen control. Microbiol Mol Biol Rev. 2007;71(3):413–451.
- Flo TH, Smith KD, Sato S, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432(7019):917–921.
- Hider RC, Kong X. Chemistry and biology of siderophores. Nat Prod Rep. 2010;27(5):637–657.
- Saoin S, Boonkrai C, Pisitkun T, et al. Expression of secreted neutrophil gelatinase-associated lipocalin in 293T cell using the inducible dual-function system. Processes. 2021;9(5):855.
- Thomas CL, Maule AJ. Limitations on the use of fused green fluorescent protein to investigate structure-function relationships for the cauliflower mosaic virus movement protein. J Gen Virol. 2000;81(Pt 7):1851–1855.
- Shaimardanova AA, Kitaeva KV, AbdrakhmanovaChernov II, et al. Production and application of multicistronic constructs for various human disease therapies. Pharmaceutics. 2019;11(11):580.
- Lee S, Kim JA, Kim HD, et al. Real-time temporal dynamics of bicistronic expression mediated by internal ribosome entry site and 2A cleaving sequence. Mol Cells. 2019;42(5):418–425.
- Albano CR, Randers-Eichhorn L, Bentley WE, et al. Green fluorescent protein as a real time quantitative reporter of heterologous protein production. Biotechnol Prog. 1998;14(2):351–354.
- Fischbach MA, Lin H, Zhou L, et al. The pathogen-associated iroA gene cluster mediates bacterial evasion of lipocalin 2. Proc Natl Acad Sci U S A. 2006;103(44):16502–16507.
- Tang X-Y, Zhou J-B, Luo F-Q, et al. Urine NGAL as an early biomarker for diabetic kidney disease: accumulated evidence from observational studies. Ren Fail. 2019;41(1):446–454.
- Pennemans V, Rigo JM, Faes C, et al. Establishment of reference values for novel urinary biomarkers for renal damage in the healthy population: are age and gender an issue? Clin Chem Lab Med. 2013;51(9):1795–1802.