
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Human papillomavirus (HPV) is the infectious etiological agent for diseases such as cervical cancer. Developing HPV vaccines is of great significance for cancer prevention. During the development of polyvalent HPV vaccines, such as 9-valent and 15-valent HPV vaccines, specifical identification of different HPV antigen is very important. The homemade HPV31 L1 VLPs was used as an immunogen to generate HPV 31 monoclonal antibodies (mAbs) through hybridoma cell culture. We generated several mAbs through hybridoma cell culture and selected two specific neutralizing antibodies, 19B2 and 19C4, which were further analyzed and characterized for binding activity, neutralizing activity, and specificity by enzyme-linked immunosorbent assays (ELISA). It is shown that both 19B2 and 19C4 could specifically recognize HPV31 L1 virus-like particles (VLPs). These two specific mAbs were further used to assemble a HPV31 antigen ELISA test kit. The assembled kit with high detection performance could be widely applied for the quantitative detection of HPV31 antigen during vaccine production as well as in the preliminary analysis of HPV31 infection.
Introduction
Cervical cancer is the second most common cancer in women globally and the principal cancer of women in the majority of developing countries, where 80% of all cases occur [Citation1]. Evidence from molecular epidemiological studies demonstrated that certain types of human papillomavirus (HPV) are the main causes of cervical cancer [Citation2]. HPV is a small, non-enveloped circular double-stranded DNA virus. The HPV genome is encapsulated in a structure with 72 capsomers composed of pentameric L1 protein [Citation3]. Over 100 HPV types have currently been identified, of which approximately 40 types are responsible for causing reproductive tract infections [Citation4].
Pathogenic HPVs are divided into two categories: low-risk types, which primarily lead to genital wart infection and illness; and high-risk types, which cause nearly all cervical cancer. However, as highlighted by Muñoz et al. [Citation1], there is still no consensus regarding the categorization of the numerous HPV types with low prevalence based on risk. Generally, 13–19 types of HPV are considered high-risk, of which 11 HPV types (16, 18, 31, 33, 35, 39, 45, 51, 52, 56 and 58) are consistently classified as high-risk [Citation1]. HPV16 together with HPV18 account for approximately 70% of cervical cancer cases, while HPV45 and HPV31 account for approximately 8% and 5% of cases, respectively [Citation5].
The native HPV virion exhibits a T = 7 icosahedral symmetrical structure [Citation6], with a capsid comprised of the major capsid protein L1 and the minor capsid protein L2. The L1 capsid protein accounts for approximately 80–90% of the total capsid protein. HPV L1 is the primary target protein of HPV vaccines with high immunogenicity [Citation7]. In the absence of the L2 protein, the L1 protein expressed in vitro can self-assemble into virus-like particles (VLPs) that closely resemble the natural virus structure and can be used to induce the production of high-titer neutralizing antibodies. The three internationally available HPV vaccines at present, including Merck & Co.’s Gardasil (HPV 6/11/16/18 quadrivalent vaccine) and Gardasil 9 (HPV 6/11/16/18/31/33/45/52/58 9-valent vaccine) as well as GlaxoSmithKline’s Cervarix (HPV 16/18 bivalent vaccine), are all developed using L1 VLPs. The L2 protein accounts for about 20% of the capsid protein and is at the center of each L1 protein pentameric capsomer [Citation8]. Although L2 protein is not the key protein for VLP formation, it plays an important role in the processes of viral infection, including attachment, endocytic cellular uptake, vesicle trafficking, nuclear entry and virus replication [Citation9]. In addition, the L2 protein plays a crucial role in viral immune escape and derivation of new genotypes [Citation8].
To further expand the protective coverage of HPV prophylactic vaccines, based on successful research and manufacture of the second-generation 9-valent vaccine, we developed a third-generation recombinant HPV 15-valent vaccine, which has obtained Investigational New Drug approval from the National Medical Products Administration of China (clinical trial approval number is 2022LP00449). This recombinant vaccine can protect against 13 high-risk virus types (16/18/31/33/35/39/45/51/52/56/58/59/68) that are etiological for malignancy, and two low-risk types (6/11), improving the rate of preventive coverage to above 95% and will become the most valent vaccine that prevents malignant tumors caused by HPV, such as cervical cancer.
While during the multiple procedures of HPV vaccines production, it is very important to control the quality of the whole process. The essential tests of this process comprise identification of HPV type, determination of antigen content, and in vitro and in vivo potency assessments. Testing methods for specific and accurate detection of HPV31 L1 VLP proteins are required during research and development. Double-antibody sandwich enzyme-linked immunosorbent assay (ELISA) which is an easy and convenient method, could be employed to fulfill all these tests during the whole process of vaccine production. The specific monoclonal antibodies (mAbs) with neutralizing activity are important components in the ELISA method, which make them an irreplaceable role in vaccine development [Citation10–12].
Thus, we cultured hybridoma cell lines capable of producing specific neutralizing monoclonal antibodies (mAbs) against HPV31 and assembled an enzyme-linked immunosorbent assay (ELISA) test kit using two selected mAbs. This assembled kit allows for rapid detection of HPV31 L1 VLPs in the HPV vaccine. More importantly, this kit could also be employed in the preliminary clinical analysis of HPV31 infection, and the mAbs could be applied in the development of more complicated immunohistochemical systems as well.
Materials and methods
Ethics statement
The plan of animal study was approved by the Animal Ethics Review Board of Beijing Health Guard Biotechnology Inc. (No. 2020-14). Animal experiments were performed in accordance with the Guide for the Care and Use of Beijing Health Guard Biotechnology Inc. The whole process of animal experiments was carried out by professional technicians and the mice were euthanized by intraperitoneal injection of sodium pentobarbital at the end of the experiment.
Animals
A total of 25 female BALB/c mice (aged 6–8 weeks, 15–20 g) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. They were housed in the Animal Laboratory Center of Beijing Health Guard Biotechnology Inc. They were reared in cages at room temperature at about 25 °C with a humidity of 50–60%, 12/12 light cycles, free access to food and water ad libitum.
Materials
Freund’s complete adjuvant was purchased from SIGMA (USA). HRP-labeled goat-anti-mouse IgG secondary antibody was obtained from Jackson Immuno Research Inc. (catalog number: 115-035-164). Culture reagents were purchased from HyClone (USA). PEG4000 was obtained from STEMCELL (USA). All other reagents used in the present work were of analytical grade and were applied without further purification.
Preparation and screening of mAbs
The method of mAb preparation has been described in a previous report [Citation13]. Briefly, the homemade HPV31 L1 VLPs as an immunogen were mixed and fully emulsified with an equal volume of Freund’s complete adjuvant (Sigma). A dose of 10 μg/mouse was immunized subcutaneously. At pre-defined time points, blood was collected, following by serum titers determined through an indirect ELISA method. Then the mice with high serum titers were subjected to a booster immunization by IP injection of the emulsified mixture of antigens and Freund’s incomplete adjuvant (Sigma) to derive immunized spleen cells. Myeloma cell suspensions were subsequently prepared according to standard instruction [Mouse Hybridoma (Sp 2/0), IMM010.51.2] (ATCC, USA) and injected into BALB/c mice. After solid tumors formed in the mice, the tumor cells were collected and fused with the immunized spleen cells (obtained from the previous operation) to derive hybridoma cells. Hybridoma cell lines with high titers were identified and screened. Then the screened cell lines were cultured in 10% fetal bovine serum (FBS) Dulbecco’s Modified Eagle Medium. The cell culture supernatants were collected and used to inoculate into mature BALB/c mice for ascites fluid collection. The supernatants of collected ascites were purified by protein G affinity chromatography columns to isolate HPV31 L1 VLP mAbs.
Indirect ELISA was utilized during primary screening to test the supernatants of the wells containing the hybridoma cells. Briefly, the homemade HPV31 VLP antigen coating solution was added to a 96-well microplate and incubated at 4 °C overnight. The blocking solution was added to the microplate followed by incubation at 37 °C for 2 h. The cell culture supernatant was diluted at a certain ratio and added to the microplate, and a negative control (NC) was set. After incubating at 37 °C for 1 h followed by washing and patting the microplate dry, the wells were subsequently incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (100 μL per well) at a ratio of 1:2000. Finally, the absorption of the supernatants at 450 nm was measured in an ELISA reader (Bio Tek, ELx808).
The selected cell cultures with positive mAb signals were further expanded for rescreening. Indirect ELISA was also utilized during rescreening. Similar to the primary screening, HPV31 L1 VLP or pentameric L1 protein was used for coating, and the cells secreting mAbs capable of recognizing two antigens simultaneously were selected. A comprehensive analysis based on the results of the experiments was performed to select the mAbs for further characterization.
Characterization of anti-HPV31 L1 VLP mAbs
Identification of antibody isotypes
An indirect ELISA method was employed to identify the IgG isotype of the antibodies obtained above. The homemade HPV31 VLP antigen coating solution (100 μL per well) was added to a 96-well microplate and incubated at 4 °C overnight. The blocking solution (200 μL per well) was added to the microplate followed by incubation at 37 °C for 2 h. The mAb was diluted at a certain ratio and added to the microplate, and a negative control (NC) was set. Binding between the mAb and VLP on the solid carrier occurred during incubation at 37 °C for 1 h. After washing and patting the microplate dry, goat anti-mouse secondary antibodies to different IgG isotypes (IgG1/IgG2a/IgG2b/IgG3/IgA/IgM/IgGλ/IgGκ) were diluted at a certain ratio, added individually to the microplate, and incubated at 37 °C for 1 h. After washing and patting the microplate dry, a colorimetric substrate solution was added and reacted at 37 °C for 10 min without light. A stop solution was added and the optical density at 450 nm (OD450) was measured.
Evaluation of antibody specificity
A microplate was coated with the homemade VLPs using 15 HPV types along with denatured VLPs, and indirect ELISA was used to evaluate the specificity of the mAbs. Native L1 VLPs were added at a ratio of 1:1 to a denaturing buffer (0.2 mol/L sodium carbonate, pH 10.6, 10 mmol/L dithiothreitol), incubated at 25 °C for 30 min, and boiled for 5 min to derive the denatured L1 VLP proteins. Native and denatured VLP proteins were diluted separately to 2 μg/mL, added to a 96-well microplate at 100 μL/well, and incubated at 4 °C overnight. After drying, the microplate was blocked by 2% bovine serum albumin (BSA) (300 μL/well) for 2 h, mixed well with antibody samples (100 μL, 0.3 μg/mL) at 25 °C, and incubated for 1 h. HRP-conjugated goat anti-mouse IgG was diluted at a ratio of 1:4000, added to the dried microplate, and incubated at 25 °C for 1 h. The microplate was washed with phosphate buffered saline (PBS) five times before adding the next colorimetric substrate solution (100 μL/well). After avoiding light reaction for 10 min, a volume of 100 μL/well stop solution was added and the final OD450 was detected.
Measurement of antibody neutralizing activity
The HPV31 pseudoviron-based neutralization assay was performed using 293FT cells following previously described methods with some modification [Citation10, Citation14]. Using a 96-well cell culture plate, 100 μL of 293FT cells (1.5 × l04 cells) per well was added, and the plate was placed in the incubator at 37 °C with 5% CO2 for 4–6 h. The purified mAbs were 4-fold serially diluted starting at a concentration of 0.5 μg/mL using Dulbecco’s modified Eagle medium (DMEM) complete medium containing 10% FBS. The diluted purified antibodies were mixed with an equal volume of diluted HPV pseudovirus (PsV) solution and incubated at 4 °C for 1 h. Subsequently, 100 μL of PsV-antibody mixture or control solution (cell control: 100 μL DMEM containing 10% FBS; PsV control: diluted antibody containing HPV) were individually added to the 96-well cell culture plate prepared 4–6 h in advance, which was then incubated continuously at 37 °C with 5% CO2 for approximately 72 h.
Next, the MiniMax module of the SpectraMax i3 Multi-Mode Microplate Reader System was employed to record the fluorescent cell count for each well, and the viral infection inhibition rate was calculated for each dilution of purified antibody. The Reed-Muench method was then used to calculate the antibody dilution factors that inhibited 50% (IC50) and 90% (IC90) of viral infection as the antibody neutralizing titers. The corresponding antibody concentrations for the antibody neutralizing titers were calculated based on the initial protein concentration of the purified antibody. The fluorescence inhibition rate was calculated using the following equation:
Serum competition experiment for mAbs in vitro by ELISA
A competitive experiment between mAbs in vitro was performed for the two selected mAbs, 19B4 and 19C2, using the ELISA method to determine if they both recognized the same VLP binding site. A volume of 100 μL per well of 2 μg/mL VLP protein was first added to a 96-well microplate and placed in a refrigerator at 4 °C overnight. Each well was blocked by adding 250 μL of 2% BSA-PBS with Tween 20 (PBST) and incubated at 37 °C for 1.5 h. The purified antibody from the 19B4 clone was diluted to 0.3 μg/mL with PBST containing 0.1% BSA solution, and the enzyme-labeled antibody from the 19C2 clone was diluted to 0.1 μg/mL with PBST containing 0.5% BSA solution. Subsequently, non-enzyme-labeled antibody and enzyme-labeled antibody were added to the plate, and the group without non-enzyme-labeled antibody was used as a non-competitive control. After incubating at 37 °C for 1 h, 300 μL per well of PBST was added and then the plate was washed five times. The color was developed by adding the commercial 3, 3′, 5, 5′ tetramethylbenzidine dihydrochloride (TMB) substrate and avoiding light. The OD450 was recorded using a BioTek 808 microplate reader. The inhibition rate of the competitive group was calculated by setting the average OD value of the non-competitive control group as 100%. A competitive experiment between mAbs and rabbit polyclonal antibodies in vitro was also performed for the two selected antibodies, 19B4 and 19C2, using the ELISA method to assess if the antigen recognition epitopes of the two mAbs were neutralizing immunodominant epitopes. A HPV31 VLP coated plate was prepared. Competitive binding between the serially diluted mAbs and a rabbit enzyme-labeled polyclonal antibody (pAb) was performed, using a group without added mAb as a non-competitive control. The competition inhibition rate was calculated, with the inhibition rate greater than 50% as the inhibition criteria.
Binding activities of 19B4 and 19C2 mAbs
A volume of 100 μL per well of HPV31 L1 VLP protein at a concentration of 5 μg/mL was added to a 96-well microplate and placed in a refrigerator at 4 °C overnight. Each well was then blocked by adding 250 μL of PBST containing 2% BSA solution and incubated at 25 °C for 1.5 h. The purified monoclonal antibodies were 10-fold serially diluted using PBST containing 0.1% BSA solution starting at 1 μg/mL. A volume of 100 μL per well serially diluted purified monoclonal antibodies was added, and PBST containing 0.1% BSA solution was used as a negative control. After incubating at 25 °C for 1.5 h, goat anti-mouse secondary antibody, diluted at a ratio of 1:2000 with PBST containing 0.5% BSA solution, was added at a volume of 100 μL per well. After incubating at 25 °C for 1 h, 300 μL per well PBST was added and the plate was washed five times. Finally, the color was formed by adding the commercial TMB substrate without light.
The OD450 was measured using a BioTek 808 microplate reader. The recorded value of the well with the sample diluent was used as a negative control, and values greater than 2-fold of the negative control average value were defined as positive. Antibody titers were calculated for the purified mAbs to achieve positive detection.
Gene sequencing of 19B4 and 19C2
The total RNA of the two monoclonal cells, 19B4 and 19C2, were individually isolated by TRIzol reagent, and then reversely transcribed to cDNA by anti-sense primers according to the instruction of the PrimeScriptTM 1st Strand cDNA synthesis kit. Then the acquired cDNA was amplified by high-fidelity PCR using universal primers for the variable regions according to the previously reported method [Citation15, Citation16]. Fragments of PCR products were inserted into T-vectors for DNA sequencing. The sequences obtained were finally translated into protein amino acid sequences, which were then subjected to alignment and analysis.
HPV31 L1 VLP test kit
Assembly of the test kit
An antibody pairing experiment for 19B4/19C2 was performed by double-antibody sandwich ELISA, and the coating antibody and capture antibody were verified. The coating antibody 19B4 was diluted to 10 μg/mL with 0.05 mol/L sodium carbonate buffer (pH 9.6). A volume of 100 μL per well of coating antibody was added in a 96-well microplate and incubated at 4 °C overnight. The coating solution was removed, and the plate was washed twice with PBST solution. Subsequently, 200 μL per well blocking solution of 3% BSA was added and incubated at 37 °C for 2 h before being washed once with PBS. A 10% sucrose solution was added and incubated at 25 °C for 1 h to preserve the antibody. After drying the plate, it was placed in an aluminum foil bag, sealed under vacuum, and stored at 4 °C.
HRP labeled 19C2 antibody (19C2-HRP) was used as a capture antibody. A volume of 100 μL per well HPV31 L1 VLPs was added in a 96-well microplate previously immobilized with coating antibody and incubated at 37 °C for 1.5 h. After rinsing the plate, 100 μL per well of 19C2-HRP (0.5 μg/mL) was added and incubated at 37 °C for 1 h. The microplate was then washed and dried followed by addition of 100 μL per well of a chromogenic substrate. The color was developed at 37 °C for 10 min avoiding light. A stop solution (50 μL/well) was then added, and the OD450 was measured.
Linearity, range, and reproducibility of the test kit
At a starting concentration of 81 μg/mL, the antigen was diluted to 27, 9, 3, 1, 0.3333, 0.1111, 0.0370, 0.0123, 0.0041 and 0.0014 μg/mL, separately. Double-antibody sandwich ELISA was performed, with 10 tests per sample. The curve of different concentrations and their corresponding OD450 was calculated with a four-parameter curve fit.
Specificity test of the test kit
The ELISA method established here was used to test the native L1 VLP of the 15 genotypes (HPV 6/11/16/18/31/33/35/39/45/51/52/56/58/59/68, 10 μg/mL in 100 μL) individually and the OD450 values were measured.
Results
Production of hybridoma cells and mAbs
According to standard experimental methods, this study developed a total of 22 anti-HPV31 L1 VLP mAbs from mouse hybridoma cells, among which two mAbs (19B4 and 19C2) were selected to assemble the HPV31 genotype test kit. To avoid any confusion, we have only included data of these two mAbs.
Characterization of mAbs to HPV31 L1 VLP
During the preliminary antibody screening, all antibody isotypes were identified using the indirect ELISA method, and our results indicated that two mAbs, 19B4 and 19C2, both belonged to the IgG1 subclass.
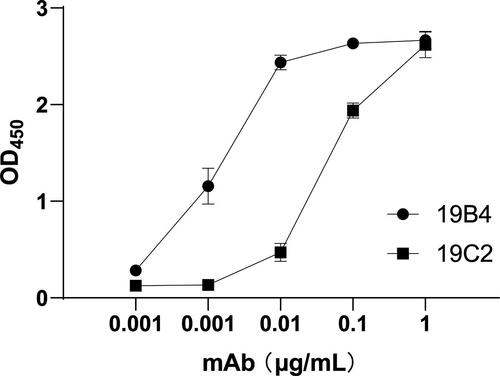
The reactivity of the mAbs to various concentrations of HPV31 L1 VLPs was determined by indirect ELISA. The binding strengths of the two mAbs to HPV31 L1 VLPs are shown in . The test results showed that both mAbs bound to HPV31 L1 VLPs at the different testing concentrations. When mAb concentration reached 0.1 μg/mL, the response signal values from both mAbs were greater than 1, which indicated that the two mAbs exhibited good reactivity and high binding affinity to HPV31 VLP.
Figure 1. The reactivity of 19B4 and 19C2 towards HPV31 L1-VLPs.
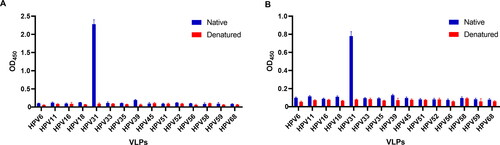
Specificity testing was then performed for the two mAbs, 19B4 and 19C2, by indirect ELISA across 15 genotypes (6/11/16/18/31/33/35/39/45/51/52/56/58/59/68). The VLP from each genotype was tested using denatured and native samples to determine if the mAbs were conformational epitope-specific antibodies. Our test results showed that the two mAbs both interacted with native HPV 31 L1 VLPs and exhibited remarkable response signals with high OD450 values, while OD450 values for other types were minimal (). These results indicated that the two mAbs, 19B4 and 19C2, did not have cross-reactivity with HPV L1 VLPs from other types, and therefore they were HPV31-specific antibodies. Compared to the experimental group using native HPV31 L1 VLPs, the OD450 values of samples reacting with denatured HPV31 L1 VLPs were considerably lower, which indicated that the two selected mAbs possessed conformational epitope specificity. To summarize, both 19B4 and 19C2 mAbs were specific to HPV31 L1 VLP.
Figure 2. Specificities of 19B4 (A) and 19C2 (B) to native or denatured HPV L1-VLPs.
Neutralizing activities for the two mAbs were detected in vitro using the well-characterized cell-based pseudovirus entry assay. The antibody dilution folds to inhibit 50% and 90% of viral infection as calculated by the Reed-Muench method were used as the antibody neutralizing titers (). These data suggested that both mAbs inhibited interaction between the pseudovirus and cells to a certain degree, thereby exhibiting neutralizing activity. A low half-maximal inhibitory concentration indicates a higher neutralizing activity of the mAb against HPV31 L1 VLPs.
Table 1. Characteristics of the HPV31 L1-VLP mAbs.
Next, competitive in vitro experiments were performed using indirect ELISA between the two mAbs as well as between the two mAbs and rabbit enzyme-labeled pAb to further determine whether the two mAbs recognized the same epitope and whether the epitopes that were recognized were immunodominant epitopes. OD450 values were recorded using a microplate reader and inhibition rates were calculated, which are shown in . Analyses of the results indicated that 19B4 and 19C2 did not compete mutually, suggesting that the two mAbs recognized distinct epitopes on HPV31 L1 VLP. Additionally, the competitive inhibition rates of 19B4 and 19C2 at the concentration of 0.1 μg/mL to rabbit pAbs were 89.2% and 92.3%, respectively. Using an inhibition rate greater than 50% as the inhibition criteria, the two mAbs both had a highly competitive capacity against rabbit pAbs. Our results demonstrated that the two mAbs identified in this study had different HPV31 L1 VLP recognition epitopes and no blocking reactions between them, and that the epitopes they recognized were both immunodominant epitopes. Thus, the two mAbs can be applied in the HPV31 test kit.
Table 2. Competition between the two mAbs, or between mAbs and rabbit pAbs in vitro.
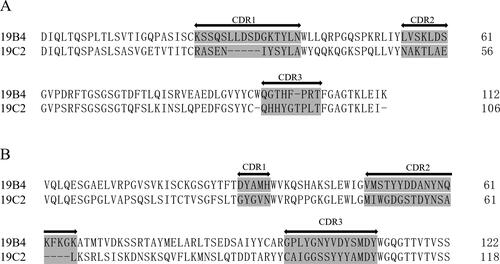
To further characterize the specificity of the two mAbs, this study performed gene sequencing for 19B4 and 19C2 (). 19B4 and 19C2 differed in the complementarity-determining regions (CDRs), shown as shaded gray in the aligned amino acid sequences in , suggesting that 19B4 and 19C2 were two distinct anti-HPV31 L1 VLP antibodies of the same isotype.
Figure 3. Alignments of amino acid sequences of the (A) light- and (B) heavy-chain regions of monoclonal antibodies (mAbs) 19B4 and 19C2. Complementarity-determining regions (CDRs) are shaded gray.
HPV31 L1 VLP test kit
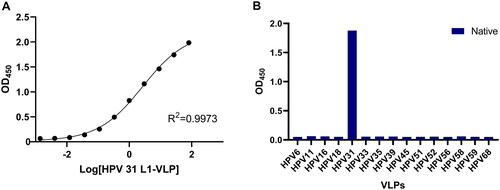
Based on the results of the pairing experiment, the test kit was assembled with 19B4 as the coating antibody and HRP-labeled 19C2 as the capture antibody. The linearity and range of the test kit were determined using a four-parameter linear fitting, and the fitted curve is displayed in . The protein concentration-response value derived for the test kit was approximately an S-shaped curve with good linearity (R square = 0.9973) in the range of 0.0014–81 μg/mL.
Figure 4. Characterization of the HPV31 L1-VLP test kit developed in this study. (A) Linearity of HPV31 test kit; (B) specificity of HPV31 test kit.
To further determine the optimal detection range and detection limit of the kit, antigen concentration was back-calculated according to the derived S-shaped curve, and the recovery ratio was calculated from these results. The range with good recovery ratios (80–120%) was determined and the linear range of detection was established as 0.1235–10 μg/mL (). The cutoff of this detection method was defined as 2-fold of the negative control (NC), and the detection limit was determined to be 0.032 μg/mL after repeated verification.
Table 3. Recovery ratio of HPV31 antigen concentration.
The specificity of the test kit was assessed by the native L1 VLP of multiple HPV types 6/11/16/18/31/33/35/39/45/51/52/56/58/59/68. Among all samples examined, only HPV31 L1 VLP generated a positive result (). It is indicated that this test kit could detect HPV31 L1 VLP with high sensitivity and without cross-reactivity with L1 VLP of other HPV types. It is also suggested that the two selected mAbs used in the test kit possessed the capacity to recognize the conformational epitopes of HPV31 L1 VLP.
Discussion
In the present study, we selected two mAbs (19B4 and 19C2) for kit assembly from 22 hybridoma cell lines capable of stably expressing anti-HPV31 L1 VLP-specific neutralizing antibodies. Several characteristics of the 19B4 and 19C2 mAbs were clarified, including antibody IgG isotypes, specificity, recognized epitopes, neutralizing capacity, binding affinity and genetic sequences. Our data indicated that 19B4 and 19C2 possessed good specificity to native HPV31 L1 VLP and recognized conformational epitopes of HPV31 L1 VLP, without cross-reactivity to L1 VLP of 14 other HPV types.
In our previously published work, we have also successfully assembled the detection kits for HPV58 and HPV16 [Citation17, Citation18]. All of these studies provide ample evidence of the generalizability of this method for construction of detected kits in the development of HPV vaccine. Generally, based on the previous studies, as for constructing the specific test kit for different HPV types, the ideal mAbs need to satisfy for the following criteria. First, the selected mAbs should specifically recognize a particular HPV antigen and the genetic sequences of each mAb should have unique specificity. Second, the two mAbs employed in the same test assay must not compete or interfere with each other; in other words, they should recognize different epitopes. The two mAbs employed in the same assay should also be able to recognize the antigen more rapidly and accurately than other mAbs; it means that they should recognize immunodominant epitopes. Third, the mAbs should have excellent binding affinities to the particular antigen and not rapidly dissociate. Last, the quality attribute of the mAbs should be the critical attribute for characterizing the quality of the test kit [Citation10].
Of all the HPV vaccines currently on the market, the 9-valent Gardasil 9 vaccine has the broadest protection range. The mAbs developed in this study not only exhibited high titer recognition of HPV31 L1 VLP for nine genotypes (the same types covered by the Gardasil 9 vaccine), but also showed good characteristics for the 15 genotypes. Therefore, it can be utilized in the detection of HPV31 VLP during the production of both 9-valent and 15-valent vaccines and in clinical detection of HPV31 infection as well. This kit could be used in routine quality control testing during HPV vaccine production.
Until now, ELISA is still a good choice to monitor the potency of HPV vaccines throughout the entire production process. Although there are many commercially available mAbs and kits for polyvalent vaccines, the use of homemade mAbs and kits is more beneficial in the long term from the point of view of controlling the cost of production and securing the entire production process.
Conclusions
We have generated two monoclonal antibodies (19B4 and 19C2) which are of high binding affinities and high titers. They were applied to the construction of a double-antibody sandwich ELISA test kit. This test kit can specifically recognize and quantitatively detect for HPV31 L1 VLPs antigens within a set of 15 genotypes (HPV 6/11/16/18/31/33/35/39/45/51/52/56/58/59/68), and thus it can be widely utilized in clinical detecting and quality control during vaccine development. Furthermore, the present study also provides a feasible strategy for screening mAbs of high titer and high affinity to apply in the diagnostics and therapeutics of HPV infection.
Authors’ contributions
Yongjiang Liu and Haijiang Zhang conceived and designed this study. Yuying Liu and Guifeng Zhang prepared the manuscript. Yao Zhang, Xuehong Wang, Zengmin Yang, Yuying Liu, Yan Wang, Minghui Zheng, Yun Liu, Jiayu Pan, Shuming Wu, and Xiao Chen conducted experiments. All authors contributed to manuscript revision and read and approved the submitted version.
Acknowledgments
We sincerely thank Professor Xiaojiang Chen at the University of Southern California for his assistance.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article.
Disclosure statement
Yuying Liu, Yao Zhang, Xuehong Wang, Zengmin Yang, Yakun Li, Yan Wang, Minghui Zheng, Yun Liu, Shuming Wu, Xiao Chen, Haijiang Zhang, and Yongjiang Liu are employees of Beijing Health Guard Biotechnology Inc.
Additional information
Funding
References
- Munoz N, Bosch FX, de Sanjose S, International Agency for Research on Cancer Multicenter Cervical Cancer Study Group, et al. Epidemiological classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348(6):518–527.
- Shah KV, Howley, PM. Field virology: papillomaviruses. Fields BN, Knipe DM, Howley PM, editors. New York: Raven Press; 1996. p. 2078–2109.
- Brendle SA, Culp TD, Broutian TR, et al. Binding and neutralization characteristics of a panel of monoclonal antibodies to human papillomavirus 58. J Gen Virol. 2010;91(Pt 7):1834–1839.
- WHO. 2022. Human papillomavirus (HPV) and cervical cancer. https://www.who.int/en/news-room/fact-sheets/detail/human-papillomavirus-(hpv)-and-cervical-cancer.
- Kabir A, Bukar M, Nggada HA, et al. Prevalence of human papillomavirus genotypes in cervical cancer in Maiduguri, Nigeria. Pan Afr Med J. 2019;33:284.
- Baker TS, Newcomb WW, Olson NH, et al. Structures of bovine and human papillomaviruses. Analysis by cryoelectron microscopy and three-dimensional image reconstruction. Biophys J. 1991;60(6):1445–1456.
- Yaegashi N, Jenison SA, Valentine JM, et al. Characterization of murine polyclonal antisera and monoclonal antibodies generated against intact and denatured human papillomavirus type 1 virions. J Virol. 1991;65(3):1578–1583.
- Wang JW, Roden RBS. L2, the minor capsid protein of papillomavirus. Virology. 2013;445(1–2):175–186.
- Tan L, Zhang TY. Cellular receptor binding and entry of human papillomavirus. Virol J. 2010;7(1):1–7.
- Brown MJ, Seitz H, Towne V, et al. Development of neutralizing monoclonal antibodies for oncogenic human papillomavirus types 31, 33, 45, 52, and 58. Clin Vaccine Immunol. 2014;21(4):587–593.
- Dias D, Van Doren J, Schlottmann S, et al. Optimization and validation of a multiplexed luminex assay to quantify antibodies to neutralizing epitopes on human papillomaviruses 6, 11, 16, and 18. Clin Diagn Lab Immunol. 2005;12(8):959–969.
- Pastrana DV, Buck CB, Pang YYS, et al. Reactivity of human sera in a sensitive, high-throughput pseudovirus-based papillomavirus neutralization assay for HPV16 and HPV18. Virology. 2004;321(2):205–216.
- Zhang Y, He Y, Li L, et al. Development and characterization of an HPV18 test kit using two novel HPV18 type-specific monoclonal antibodies. Diagn Pathol. 2018;13(1):55.
- Sehr P, Rubio I, Seitz H, et al. High-throughput pseudovirion-based neutralization assay for analysis of natural and vaccine-induced antibodies against human papillomaviruses. PLoS One. 2013;8(10):e75677.
- Orlandi R, Güssow DH, Jones PT, et al. Cloning immunoglobulin variable domains for expression by the polymerase chain reaction. Proc Natl Acad Sci U S A. 1989;86(10):3833–3837.
- Carter P, Presta L, Gorman CM, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A. 1992;89(10):4285–4289.
- Liu YY, Zhang Y, Zhang GF, et al. Development of HPV58 type-specific antibodies and detection kit. Prep Biochem Biotechnol. 2023;53(2):223–229.
- Liu YY, Zhang Y, Zhang GF, et al. Development and characterization of anti‐HPV16 monoclonal antibodies for assembly of an HPV16 detection kit. Biotechnol Appl Biochem. 2022. DOI: 10.1002/bab.2384.