
Abstract
Leber’s hereditary optic neuropathy (LHON) is a rare maternally inherited disease caused by mutations in mitochondrial DNA (mtDNA) genes encoding subunits of complex I in the mitochondrial respiratory chain. The most common mutations causing LHON are G11778A, G3460A and T14484C, but there are also several less common mutations. LHON presents as acute or subacute bilateral visual loss, usually affecting young males. The aim of this study was to assess the clinical symptomatology and genetic analysis of Bulgarian patients with LHON. Twenty-two patients were diagnosed with LHON based on clinical evaluation and genetic examination (17 males and 5 females); 12 of them were previously reported, while 8 males and 2 females are newly diagnosed. A full neuroophthalmologic and genetic examination was performed. Eight patients had a family history of LHON, while 14 were isolated cases. The age at onset ranged from 3 to 43 years, and visual acuity ranged from counting fingers to 0.9. Genetic testing revealed various mutations, including a rare mutation G3635A in MT-ND1 in five affected members of one pedigree and digenic inheritance of G11778A and T14484C in three individuals from a different family. A variant m.15988A > G in the mitochondrial gene MT-TP with a high level of heteroplasmy was found in one patient. In addition to the most common mutations causing LHON, our patients also had rare mutations. These results suggest that genetic analysis of the entire mtDNA sequence is recommended in cases with strong clinical suspicion of LHON, since new rare mtDNA pathogenic variants are being identified.
Introduction
Leber’s hereditary optic neuropathy (LHON), as the name suggests, was first described by Theodor Leber, a German ophthalmologist, in 1871, who observed an acute onset of vision loss in both eyes in young men of four families. Initially, an X-linked mode of inheritance of the disease was suggested. In 1988, LHON was found to belong to the group of mitochondrial diseases that are inherited from the maternal side alone and are due to primary mutations in the mitochondrial genome [Citation1]. Mutations in the genes encoding MT-ND1, MT-ND4, MT-ND4L and MT-ND6 subunits of complex I in the mitochondrial respiratory chain cause LHON. The three most common point mutations in the mitochondrial genome that cause the disease in 85–90% of cases, are G11778A, T14484C and G3460A, although other relatively rarer mutations are also known [Citation2–5]. As a consequence of these mutations in the mitochondrial genome, ATP production is reduced and the number of free radicals increases, leading to retinal ganglion cell dysfunction and apoptosis, and consequently, to visual loss [Citation2,Citation6]. Not every carrier of a gene mutation develops the disease; furthermore, male carriers have a 4–5-fold higher risk of developing the disease than female carriers for unknown reasons [Citation7]. The ratio of ‘affected’ to ‘normal’ mitochondria, expressed as a percentage, is also relevant. The presence of both ‘affected’ and ‘normal’ mitochondria is termed heteroplasmy, whereas in the presence of only ‘affected’ mitochondria, the condition is termed homoplasmy. Despite the higher risk of developing the disease in homoplasmic carriers, there are known homoplasmic carriers who are completely healthy [Citation8]. The presence of additional exogenous factors and toxic substances, such as smoking, alcohol consumption, certain anti-viral, anti-malarial drugs, antibiotics, anti-arrhythmic, anti-carcinogenic, anti-hypertensive drugs, anti-convulsants, stress and vitamin B12 deficiency is also important for the occurrence of the disease in the carriers [Citation9–12]. A protective role of female sex hormones [Citation13] or some specificity of female metabolism [Citation14,Citation15] has been suggested for the lower morbidity in women.
The prevalence of the disease in the population in northern European countries ranges from 1:31,000 to 1:50,000 [Citation4,Citation16], and the G11778A mutation is responsible for the onset of the disease in approximately 70–75% of LHON patients from North America and Europe [Citation17,Citation18] and is associated with a more severe clinical course [Citation19].
The clinical characteristics of LHON include bilateral, painless acute or subacute loss of vision in individuals of young age, predominantly young males. The disease most commonly debuts in the second and third decades of life, but a later age of onset is possible, and extremely rare cases with onset even in the seventh/eighth decades have been described [Citation20,Citation21]. More commonly, the vision of one eye is affected, followed by involvement of the second eye within 1–2 months. Severe bilateral visual loss is characteristic, in the majority of cases reaching the severity of counting fingers in front of the eye, and a large central or centrocecal scotoma is detected when kinetic or static perimetry is possible. Colour perception in the red-green spectrum is also impaired. Pupillary responses to light are relatively preserved. In the acute stage, specific ophthalmoscopic changes such as optic nerve papillary hyperemia, peripapillary neurofibrillary retinal oedema, retinal telangiectasias, and marked tortuosity of retinal vessels are observed. After the acute phase, bilateral optic nerve atrophy forms within a few weeks or months. In the majority of patients, visual acuity remains definitively low. In some patients, visual function may improve, for example in a higher percentage of T14484C mutation carriers [Citation3]. As a rule, bilateral visual loss persists unchanged in most patients with LHON, and they remain with reduced visual acuity [Citation22,Citation23].
The diagnosis of LHON requires evaluation of anamnestic data regarding the characteristics of visual disturbances, possible family history on the part of the mother; complete neuro-ophthalmological examination including determination of visual acuity, kinetic or automated threshold static perimetry, anterior segment biomicroscopy, pupillary reaction study, colour perception, ophthalmoscopy; if necessary, optical coherence tomography and electrophysiological tests (electroretinography and visual evoked potentials), and molecular genetic testing conclusive of the disease. Magnetic resonance imaging (MRI) of the brain is performed to exclude optic neuropathies of other genesis, liquor examination if necessary, autoantibody testing to exclude infectious or vasculitic aetiology of the optic nerve lesion, etc.
Although LHON is a rare disease, it is common in neurological and ophthalmological practice and often goes unrecognized and misdiagnosed. This determined our aim to summarize the results of clinical and genetic studies in verified Bulgarian patients with LHON, as a continuation of our previous studies [Citation24,Citation25].
Subjects and methods
Patients
We present 22 patients with clinical symptoms of bilateral visual impairment and genetically confirmed LHON; 12 of them were previously reported [Citation24–26], while 8 males and 2 females are newly diagnosed. All of the patients were examined at the Clinic of Neurological Diseases, University Hospital “Aleksandrovska” (Sofia, Bulgaria). The genetic tests were performed at the Genome Diagnostics Laboratory, Molecular Medicine Center, Medical University of Sofia and at the Genetic Medico-Diagnostic Laboratory “Genica”, Sofia, between March 2017 and November 2022. The gender and age distribution were as follows: 17 males and 5 females, and their age at onset of visual disturbance ranged from 3 to 43 years (mean age = 19.1 years).
The patients underwent a complete neuro-ophthalmological examination including determination of best corrected visual acuity, kinetic or automated threshold static perimetry, anterior segment biomicroscopy, pupillary response examination, colour perception, direct ophthalmoscopy in medicated mydriasis and optical coherence tomography.
Brain MRI was performed in 18 patients. In all patients, the diagnosis of LHON was made definitively after molecular genetic testing and demonstration of a disease-specific mutation in the mitochondrial genome.
Analysis of mitochondrial DNA
All study participants or their parents/legal guardians provided written informed consent.
Peripheral blood samples were collected from participants and genomic DNA was isolated from leukocytes using Chemagic DNA blood 10 k kit H1 and Chemagen Magnetic Separation Module (PerkinElmer®, Waltham, MA) according to the manufacturer’s protocol, or using a standard salting-out method. Genetic analysis was performed by Sanger sequencing of 37 mitochondrially encoded genes (MT-RNR1, MT-RNR2, MT-TA, MT-TR, MT-TN, MT-TD, MT-TC, MT-TE, MT-TQ, MT-TG, MT-TH, MT-TI, MT-TL1, MT-TL2, MT-TK, MT-TM, MT-TF, MT-TP, MT-TS1, MT-TS2, MT-TT, MT-TW, MT-TY, MT-TV, MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND4L, MT-ND5, MT-ND6, MT-CYB, MT-CO1, MT-CO2, MT-CO3, MT-ATP6, MT-ATP8) or of targeted LHON mutations. DNA fragments were amplified by polymerase chain reaction (PCR) with specific primers. The primers were designed in-house, by using the UCSC In-Silico PCR online software (https://genome.ucsc.edu/cgi-bin/hgPcr) on the basis of Ramos et al. [Citation27] (). PCR was performed by using 80 ng of DNA in a reaction containing 5 mmol/L dNTPs mixture, 10× Prime Taq buffer (22.5 mmol/L MgCl2), 5 U/μL Thermostable Prime Taq polymerase and 10 pmol/μL reverse and forward primers.
Table 1. Validated primers to amplify the complete mtDNA.
Cycle conditions were 5 min at 94 °C for the initial denaturation, followed by 30 cycles of 94 °C for 40 s, 60 °C for 40 s, 72 °C for 1 min and 5 min at 72 °C for the final extension (ABI GeneAmp PCR System 2700, Applied Biosystems, Foster City, CA).
Sanger sequencing of the targeted genes was performed with the Big Dye® Terminator cycle sequencing kit v.3.1 (Applied Biosystems). Sequencing analysis was conducted using standard electrophoresis conditions on a 36 cm capillary array with POP7 polymer (Applied Biosystems). Capillary electrophoresis was performed using ABI 3500XL and ABI 3130 genetic analyzers (Applied Biosystems). Sequencing profiles were interpreted with the Sequencing Analysis v.5.1.1 software.
Results
In our cohort of 22 patients with LHON, the majority of patients were male (17 patients), with 5 affected females, that is, the number of affected male individuals was about three times higher.
In the 22 patients studied, the onset of the first subjective complaints of ‘decrease in vision’, ‘blurring in front of one or both eyes’ was in the age range from about 3 to 43 years (), with the mean age of onset of the first subjective complaints in the group being 19.1 years.
Table 2. Distribution of patients with LHON (n = 22) according to age of onset of first symptoms of the disease.
The visual symptoms developed acutely within a few days in 8 patients from our group, and subacutely over a period of 1–2 months in 10 patients. In three cases with onset at around three years of age and in one case with onset at around 6 years of age, the exact onset of visual disturbances cannot be determined precisely, nor can the initial course of the disease. In three of these cases, parents’ observations of ‘reduced vision’ motivated them to carry out ophthalmological/neuro-ophthalmological examinations in their children, followed by confirmation of the diagnosis.
In 15 patients with LHON, the typical and more frequent pattern of initially affecting one eye and, within 1–3 months or more, affecting the second eye was present. In three patients, a rarer pattern of simultaneous worsening of vision in both eyes was observed.
In three patients with onset at around 3 years of age and one patient with onset at around 6 years of age, involvement of both eyes was detected, but it is not possible to determine with certainty whether one eye was initially affected or both eyes were affected simultaneously.
In four LHON patients, neuro-ophthalmological examination was performed in the acute stage of the disease, within 1–1.5 months after the onset of clinical symptoms, and in the remaining 18 patients, months or years after the initial onset of visual disturbances.
In all patients – in the acute or post-acute stage, bilateral decrease in visual acuity was observed. The distribution of visual acuity in the group of patients is presented in .
Table 3. Distribution of visual acuity in patients with LHON (n = 22).
Visual acuity varied from very low, below 0.05, which is by definition practical blindness, to 0.9 (1 affected person), which is close to normal visual acuity.
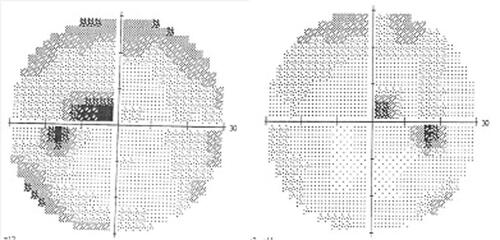
There were also typical perimetric findings in the patients: bilateral central/cecocentral scotomas or, in milder cases, paracentral scotomas (). Biomicroscopic examination of the anterior eye segment in patients did not reveal any abnormal findings. In all examined patients, the pupillary reactions were relatively preserved without a manifest afferent pupillary defect, which supports the differential diagnosis with other lesions of the optic nerve, such as optic neuritis. Colour perception was impaired in the patients, and they usually reported impaired colour vision as one of the first manifestations of the disease along with decreased visual acuity.
Figure 1. Bilateral central scotomas in the visual field of a patient, 28 years old, with LHON.
In the four patients that we examined in the acute stage, there was a typical ophthalmoscopic picture of hyperemia of the optic disc, tortuosity of the retinal vessels and often the presence of telangiectasia ().
Figure 2. Photograph of the fundus of the eye of a patient, age 16 years, with LHON in the acute stage, showing mild hyperemia of the optic disc and tortuosity of the retinal vessels [Citation24].
![Figure 2. Photograph of the fundus of the eye of a patient, age 16 years, with LHON in the acute stage, showing mild hyperemia of the optic disc and tortuosity of the retinal vessels [Citation24].](/cms/asset/78c12d42-87c0-4ff3-bac6-f5dc95344ff4/tbeq_a_2255073_f0002_c.jpg)
After a period of weeks or months, similar ophthalmoscopic symptoms specific to the acute stage were no longer observed, ‘disappearing’, as hyperemia of the optic disc is replaced by a picture of atrophy of the optic nerve. Bilateral optic atrophy persists as a definitive finding in the patients.
Eighteen of our patients were neuro-ophthalmologically examined after the acute stage of the disease, and typical ophthalmoscopic findings were associated with bilateral optic atrophy.
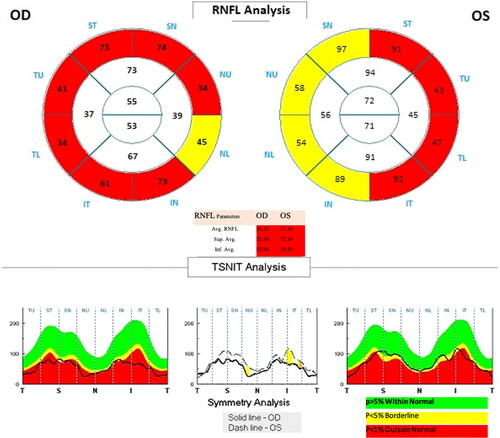
In patients with LHON who underwent optical coherence tomography with programs for analysis of the RNFL (retinal nerve fibre layer) and GCC (ganglion cell complex), there was typical ‘thinning’ of the RNFL and GCC, an expression of axonal loss and degeneration of retinal ganglion cells ().
Figure 3. Optical coherence tomography in a patient with LHON – data on manifest ‘thinning’ of the retinal neuro-fibre layer in both eyes.
Reduced visual acuity was the dominant symptom in all of our patients with LHON. The neurological status did not show the presence of added neurological symptoms except for one patient (10 years old), who had the following symptoms in addition to bilateral optic atrophy: paralysis of vertical version eye movements in the up/down gaze, preservation of horizontal version eye movements with bilateral horizontal gaze-evoked nystagmus and a slightly expressed myopathic syndrome with bilateral ptosis of the eyelids. Similar symptoms with impairment of extraocular muscle functions (chronic progressive external ophthalmoplegia) and myopathic syndrome are common in mitochondrial diseases and were added to the manifest visual impairments in this case.
Among the remaining 21 patients with LHON, we did not find additional neurological symptoms such as peripheral neuropathy, tremor, myopathy, external ophthalmoplegia and others. MRI of the brain was performed on 18 patients, with normal findings observed in 14 patients, including no changes in both optic nerves traced along their retrobulbar and intracranial segments. In three patients with LHON, single non-specific gliotic foci in white matter were observed, hyperintense in T2 and FLAIR, and in one patient, a mild thickening of both optic nerves was also observed, interpreted as an indication of inflammation (neuritis). In the last patient, MRI of the brain was performed during the acute phase of the disease, within 1 month of the onset of the first visual disturbances. Gliotic changes were found in the mesencephalic tegmentum and lamina quadrigemina of one patient. According to literature data, MRI of the brain usually reveals normal findings or occasionally lesions in white matter, respectively hyperintense lesions along the course of the optic nerves in patients with LHON.
Genealogical analysis in our patients showed familial occurrence in 8 patients belonging to 2 unrelated families, while the remaining 14 patients were sporadic (isolated) cases in their families. Molecular genetic testing confirmed the presence of mutations that cause the disease ().
Table 4. Distribution of LHON patients (n = 22) according to the type of mutation causing the disease.
Interestingly, we found the G11778A mutation in MT-ND4 in combination with the T14484C mutation in MT-ND6 in three patients from one family in the studied group. All three affected individuals showed homoplasmy.
Among the other five affected individuals belonging to another family, a relatively rare mutation causing LHON was found. The family has five affected individuals: two brothers, their mother, the mother’s sister and her son. Тhe first four members were previously diagnosed and reported [Citation24]. Meanwhile a new family member was diagnosed: the mother’s sister’s son. Sequencing analysis of targeted mitochondrial genes in the mother and both of her sons showed the presence of the G3635A mutation, p.S110N in MT-ND1, causing LHON ().
Figure 4. A family with LHON with five affected individuals (four of them previously reported [Citation24]) with the G3635A, p. S110N mutation in MT-ND1.
![Figure 4. A family with LHON with five affected individuals (four of them previously reported [Citation24]) with the G3635A, p. S110N mutation in MT-ND1.](/cms/asset/f908893d-aa73-49fc-973d-1bf1c3614e7b/tbeq_a_2255073_f0004_b.jpg)
It is of interest that this mutation is relatively rare in the European population, but is found in Russian families of Siberian origin as well as in the Chinese ethnicity [Citation28–30]. This family exhibits relatively mild visual impairment, with one of the patients having visual acuity of 0.9, which is close to normal visual acuity. In the studied LHON group, we found the three most common mutations for the European population, as well as the less common G3635A mutation in MT-ND1, which is found in Asian ethnicities. The presence of more than one LHON-related mitochondrial genome mutation was also observed in one of the studied families. In one patient, with onset of visual symptoms at the age of 31, a new genetic variant m.15988A > G with high level of heteroplasmy was detected for the first time and was described in our previous report [Citation26]. Based on the clinical diagnosis, mitochondrial genome sequencing was performed on the patient. The molecular genetic analysis showed the presence of a genetic variant in the mitochondrial genome A15988G in high level of heteroplasmy in the MT-TP gene encoding the tRNA for proline. The patient exhibited relatively mild clinical symptoms with moderately reduced visual acuity and a relatively mild perimetric defect.
Discussion
LHON is a rare maternally inherited disease caused by point mutations in mitochondrial DNA. The disease affects predominantly males, which is a well-known fact in the literature with various hypothetical explanations for this phenomenon [Citation2,Citation3,Citation7,Citation13–15,Citation23].
According to literature data, involvement of the second eye usually occurs within 1–3 months or sometimes over a longer period of time, and both eyes are affected simultaneously in around 20–25% of patients [Citation7,Citation31,Citation32].
The classical symptoms of the acute phase of LHON are: peripapillary telangiectatic microangiopathy in about 30–60% of eyes, oedema of the neuro-fibrillary layer around the optic disc and absence of fluorescein leakage from the papilla of the optic nerve or papillary area, which distinguishes it from stasis papilla, for example, Refs. [Citation33–35]. These characteristic changes in the fundus may be discreetly manifested or even absent in about 20% of patients.
The patient who showed neurological symptoms comes from a family with early onset of visual impairment (around the age of three) and the presence of other affected individuals. The patient’s mother and younger brother, who have a clear picture of bilateral optic atrophy, and other relatives with ‘visual disturbances’ are also reported, but they have not been clinically examined. The mother and her two sons (10 and 7 years old) were shown to have a homoplasmic carrier status for mutations G11778A, p.R340H in MT-ND4 and T14484C, p.M64V in MT-ND6, causing LHON. Visual impairments are the cardinal and only symptom in patients with LHON, but there are reports of individual cases where visual symptoms may be associated with added clinical symptoms such as conductive heart defects, skeletal abnormalities, discrete or more manifest neurological symptoms, and in the presence of more severe added symptoms, the condition is defined as ‘LHON plus syndrome’ [Citation2,Citation22]. Such neurological symptoms include postural tremor, peripheral neuropathy, myopathy, extrapyramidal disorders and sometimes symptoms resembling the clinical picture of multiple sclerosis [Citation36,Citation37]. The presence of other neurological symptoms added to bilateral optic atrophy determines the case of the 10-year-old patient as ‘LHON plus syndrome’.
The obtained results show that out of the three primary mutations, G11778A in MT-ND4, T14484C in MT-ND6 and G3460A in MT-ND1, which are responsible for the disease in around 90% of cases among the European population, we find two: G11778A and G3460A in 12 of our sporadic cases. It is known that the G11778A mutation is the most common among the European population and is associated with the relatively most severe clinical picture of visual loss [Citation2,Citation3,Citation23]. In support of this observation, 8 out of a total of 10 patients with visual acuity below 0.05 (practical blindness) in our study have the G11778A mutation in MT-ND4.
Unlike the G11778A mutation, which is associated with severe visual impairment, the T14484C mutation determines a more favourable course of the disease for the patient, with possibilities for improvement of visual acuity after the initial visual loss. In the three affected individuals from this family, the visual acuity is around 0.09–0.1, indicating that there is a relatively moderately expressed reduction in vision. It is possible that this relatively milder visual impairment is related to the T14484C mutation, which modifies the effect of the G11778A mutation. There are data in the literature about families (of Chinese ethnicity) in which more than one mutation in mitochondrial genes associated with LHON have been observed [Citation38].
In patients with the mtDNA mutation C4640A, p.I57M, there is a homoplasmic transversion from C to A at nucleotide 4640 in the ND2 gene, representing a known missense mutation that leads to the development of LHON.
LHON is primarily caused by the well-known primary mutations in mtDNA in most populations, although a small percentage of cases result from rarer mutations. Therefore, it is recommended to perform a comprehensive examination of the mitochondrial genome in cases of suspected LHON.
Until recently, pharmacological therapy was not applied to patients with LHON. ‘Auxiliary optical aids for low vision’ such as magnifying glasses and telescopic glasses were recommended for these patients. Antioxidant agents that ‘bypass’ complex I of the mitochondrial respiratory chain may prevent the apoptosis of retinal ganglion cells associated with LHON. One such antioxidant agent is the short-chain synthetic benzoquinone Idebenone, which acts as an electron transfer directly to complex III of the mitochondrial respiratory chain, thereby improving ATP synthesis and reactivating retinal ganglion cells. Randomized, multicenter, double-blind and placebo-controlled studies have shown good efficacy and safety of oral Idebenone [Citation39–44].
In recent years, clinical trials have been conducted on gene therapy based on intravitreal application of adeno-associated viral vectors encoding the human wild type of the ND4 gene, with the aim of restoring complex I of the mitochondrial respiratory chain and preventing degeneration of retinal ganglion cells in patients with LHON. Promising results have been reported regarding the efficacy of the treatment and the safety profile [Citation45–49].
These treatment options underscore the need for recognition and early diagnosis of LHON.
Authors’ contributions
S.C.: conception, study design, execution, acquisition of data, analysis and interpretation, writing the manuscript; B.Z.: execution, acquisition of data; K.K.: study design, execution, acquisition of data, analysis and interpretation; K.M.: acquisition of data, analysis; S.A.: execution, acquisition of data, analysis; T.T.: execution, acquisition of data, analysis; V.H.: acquisition of data, analysis and interpretation, writing and revising the manuscript; I.T.: conception, study design, execution, revision and critical review of the manuscript; R.K.: conception, study design, revision and critical review of the manuscript; A.T.: conception, study design, execution, acquisition of data, analysis and interpretation. A.O.: conception, study design, execution, critical review of the manuscript. All authors have read and approved the final version of the article.
Ethics statement
An informed consent form was signed by all participants and their relatives. The present study adhered to the principles of the Declaration of Helsinki.
Disclosure statement
All authors declare that they have no relevant financial or nonfinancial interests to disclose regarding the publication of this article.
Data availability statement
The anonymized data that support the findings of this study are available on request from the corresponding author [S.C.]. The data are not publicly available due to privacy or ethical restrictions.
Additional information
Funding
References
- Wallace D, Singh G, Lott M, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242(4884):1–9. doi: 10.1126/science.3201231.
- Fraser J, Biousse V, Newman N. The neuro-ophthalmology of mitochondrial disease. Surv Ophthalmol. 2010;55(4):299–334. doi: 10.1016/j.survophthal.2009.10.002.
- Yu-Wai-Man P, Turnbull D, Chinnery P. Leber hereditary optic neuropathy. J Med Genet. 2002;39(3):162–169. doi: 10.1136/jmg.39.3.162.
- Yu-Wai-Man P, Griffiths G, Brown D, et al. The epidemiology of Leber hereditary optic neuropathy in the North East of England. Am J Hum Genet. 2003;72(2):333–339. doi: 10.1086/346066.
- Bhate M, Kulkarni S, Nalawade R, et al. Clinical and genetic profile of Leber’s hereditary optic neuropathy in a cohort of patients from a tertiary eye care center. J Pediatr Ophthalmol Strabismus. 2022;59(5):344–349. doi: 10.3928/01913913-20220124-02.
- Howell N. Leber hereditary optic neuropathy: respiratory chain dysfunction and degeneration of the optic nerve. Vision Res. 1998;38(10):1495–1504. doi: 10.1016/s0042-6989(97)00444-6.
- Riordan-Eva P, Sanders M, Govan G, et al. The clinical features of Leber’s hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation. Brain. 1995;118(Pt 2):319–337. doi: 10.1093/brain/118.2.319.
- Kerrison J, Newman N. Clinical spectrum of Leber’s hereditary optic neuropathy. Clin Neuroscience. 1997;4(5):295–301.
- Carelli V, d‘Adamo P, Valentino ML, et al. Parsing the differences in affected with LHON: genetic versus environmental triggers of disease conversion. Brain. 2016;139(Pt 3):e17. doi: 10.1093/brain/awv339.
- Pott J, Wong K. Leber’s hereditary optic neuropathy and vitamin B12 deficiency. Graefes Arch Clin Exp Ophthalmol. 2006;244(10):1357–1359. doi: 10.1007/s00417-006-0269-7.
- Sadun A, Carelli V, Salomao S, et al. Extensive investigation of a large Brazilian pedigree of 11778/haplogroup J Leber hereditary optic neuropathy. Am J Ophthalmol. 2003;136(2):231–238. doi: 10.1016/s0002-9394(03)00099-0.
- Yu-Wai-Man P, Griffiths P, Chinnery P. Mitochondrial optic neuropathies—disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011;30(2):81–114. doi: 10.1016/j.preteyeres.2010.11.002.
- Giordano C, Montopoli M, Perli E, et al. Oestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathy. Brain. 2011;134, 1(Pt 1):220–234. doi: 10.1093/brain/awq276.
- Blaak E. Gender differences in fat metabolism. Curr Opin Clin Nutr Metab Care. 2001;4(6):499–502. doi: 10.1097/00075197-200111000-00006.
- Power M, Schulkin J. Sex differences in fat storage, fat metabolism, and the health risks from obesity: possible evolutionary origins. Br J Nutr. 2008;99(5):931–940. doi: 10.1017/S0007114507853347.
- Puomila A, Hämäläinen P, Kivioja S, et al. Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland. Eur J Hum Genet. 2007;15(10):1079–1089. doi: 10.1038/sj.ejhg.5201828.
- Mackey D, Oostra R, Rosenberg T, et al. Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy. Am J Hum Genet. 1996;59(2):481–485.
- Yu-Wai-Man P, Votruba M, Burte F, et al. A neurodegenerative perspective on mitochondrial optic neuropathies. Acta Neuropathol. 2016;132(6):789–806. doi: 10.1007/s00401-016-1625-2.
- Newman N, Carelli V, Taiel M, et al. Visual outcomes in Leber hereditary optic neuropathy patients with the m. 11778G > A (MTND4) mitochondrial DNA mutation. J Neuroophthalmol. 2020;40(4):547–557. doi: 10.1097/WNO.0000000000001045.
- Buchan J, Ong C, Dabbs T. Acute leber hereditary optic neuropathy in a 73-year-old man. Eye (Lond). 2007;21(6):859–860. doi: 10.1038/sj.eye.6702729.
- Dimitriadis K, Leonhardt M, Yu-Wai-Man P, et al. Leber’s hereditary optic neuropathy with late disease onset: clinical and molecular characteristics of 20 patients. Orphanet J Rare Dis. 2014;9(1):158. doi: 10.1186/s13023-014-0158-9.
- Kirkman M, Korsten A, Leonhardt M, et al. Quality of life in patients with leber hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2009;50(7):3112–3115. doi: 10.1167/iovs.08-3166.
- Yu-Wai-Man P, Votruba M, Moore A, et al. Treatment strategies for inherited optic neuropathies: past, present and future. Eye (Lond). 2014;28(5):521–537. doi: 10.1038/eye.2014.37.
- Cherninkova S, Zaharova B, Sareva R, et al. Clinical symptoms and genetic studies in patients with Leber’s hereditary optic neuropathy. Bulg Neurol. 2016;17(3):226– 233.
- Cherninkova S, Zaharova B, Saraeva R, et al. Leber’s hereditary optic neuropathy in bulgarian patients. C R Acad Bulg Sci. 2020;73(1):125–132.
- Cherninkova S, Atemin S, Todorov T, et al. A case of Leber’s hereditary optic neuropathy with a new genetic variant m.15988A > G in the mitochondrial genome. Bulg Neurol. 2018;19(2):62–67.
- Ramos A, Santos C, Alvarez L, et al. Human mitochondrial DNA complete amplification and sequencing: a new validated primer set that prevents nuclear DNA sequences of mitochondrial origin co-amplification. Electrophoresis. 2009;30(9):1587–1593. PMID: 19350543. doi: 10.1002/elps.200800601.
- Volodko N, L’vova M, Starikovskaya E, et al. Spectrum of pathogenic mtDNA mutations in Leber’s hereditary optic neuropathy families from Siberia. Russian J Genet. 2006;42(1):89–97.
- Yang J, Zhu Y, Tong V, et al. Confirmation of the mitochondrial ND1 gene mutation G3635A as a primary LHON mutation. Biochem Biophys Res Commun. 2009;386(1):50–54. doi: 10.1016/j.bbrc.2009.05.127.
- Bi R, Zhang A, Jia X, et al. Complete mitochondrial DNA genome sequence variation of Chinese families with mutation m.3635G > a and leber hereditary optic neuropathy. Mol Vis. 2012;18:3087–3094.
- Carroll WM, Mastaglia FL. Leber’s optic neuropathy: a clinical and visual evoked potentials study of affected and asymptomatic members of a six generation family. Brain. 1979;102(3):559–580. doi: 10.1093/brain/102.3.559.
- Nikoskelainen E, Hoyt W, Nummelin K. Fundus findings in Leber’s hereditary optic neuroretinopathy. Ophthalmic Paediatr Genet. 1985;5(1-2):125–130. doi: 10.3109/13816818509007866.
- Nikoskelainen E, Hoyt W, Nummelin K. Ophthalmoscopic findings in Leber’s hereditary optic neuropathy. I. Fundus findings in asymptomatic family members. Arch Ophthalmol. 1982;100(10):1597–1602. doi: 10.1001/archopht.1982.01030040575003.
- Nikoskelainen E, Hoyt W, Nummelin K. Ophthalmoscopic findings in Leber’s hereditary optic neuropathy. II. The fundus findings in the affected family members. Arch Ophthalmol. 1983;101(7):1059–1068. doi: 10.1001/archopht.1983.01040020061011.
- Harding A, Sweeney M, Govan G, et al. Pedigree analysis in Leber hereditary optic neuropathy families with a pathogenic mtDNA mutation. Am J Hum Genet. 1995;57(1):77–86.
- McFarland R, Chinnery P, Blakely E, et al. Homoplasmy, heteroplasmy, and mitochondrial dystonia. Neurology. 2007;69(9):911–916. doi: 10.1212/01.wnl.0000267843.10977.4a.
- Martikainen M, Ng Y, Gorman G, et al. Clinical, genetic, and radiological features of extrapyramidal movement disorders in mitochondrial disease. JAMA Neurol. 2016;73(6):668–674. doi: 10.1001/jamaneurol.2016.0355.
- Shu L, Zhang Y, Huang X, et al. Complete mitochondrial DNA sequence analysis in two Southern Chinese pedigrees with Leber hereditary optic neuropathy revealed secondary mutations along with the primary mutation. Int J Ophthalmol. 2015;5(1):28–31.
- Klopstock T, Yu-Wai-Man P, Dimitriadis K, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain. 2011;134(Pt 9):2677–2686. doi: 10.1093/brain/awr170.
- Klopstock T, Metz G, Yu-Wai-Man P, et al. Persistence of the treatment effect of idebenone in Leber’s hereditary optic neuropathy. Brain. 2013;136(Pt 2):e230.doi: 10.1093/brain/aws279.
- Lyseng-Williamson K. Idebenone: a review in Leber’s hereditary optic neuropathy. Drugs. 2016;76(7):805–813. doi: 10.1007/s40265-016-0574-3.
- Carelli V, Carbonelli M, de Coo I, et al. International consensus statement on the clinical and therapeutic management of Leber hereditary optic neuropathy. J Neuroophthalmol. 2017;37(4):371–381. doi: 10.1097/WNO.0000000000000570.
- Catarino C, von Livonius B, Priglinger C, et al. Real-world clinical experience with idebenone in the treatment of Leber hereditary optic neuropathy. J Neuroophthalmol. 2020;40(4):558–565. doi: 10.1097/WNO.0000000000001023.
- van Everdingen JAM, Pott JWR, Bauer NJC, et al. Clinical outcomes of treatment with idebenone in leber’s hereditary optic neuropathy in The Netherlands: a national cohort study. Acta Ophthalmol. 2022;100(6):700–706. doi: 10.1111/aos.15153.
- Newman N, Yu-Way-Man P, Carelli V, et al. Efficacy and safety of intraviteral gene therapy for Leber hereditary optic neuropathy treated within 6 months of disease onset. Ophthalmology. 2021;128(5):649–660. doi: 10.1016/j.ophtha.2020.12.012.
- Sahel J, Newman N, Yu-Way-Man P, et al. Gene therapies for the treatment of Leber hereditary optic neuropathy. Int Ophthalmol Clin. 2021;61(4):195–208. doi: 10.1097/IIO.0000000000000364.
- Carelli V, Newman NJ, Yu-Wai-Man P, the LHON Study Group., et al. Indirect comparison of lenadogene nolparvovec gene therapy versus natural history in patients with Leber hereditary optic neuropathy carrying the m.11778G > a MT-ND4 mutation. Ophthalmol Ther. 2023;12(1):401–429. doi: 10.1007/s40123-022-00611-x.
- Carper MG, Henderson AD. Updated review of Leber hereditary optic neuropathy. Curr Treat Options Neurol. 2022;24(9):441–452. doi: 10.1007/s11940-022-00729-0.
- Yu-Wai-Man P, Newman NJ, Carelli V, LHON REALITY Study Group., et al. Natural history of patients with leber hereditary optic neuropathy-results from the REALITY study. Eye (Lond). 2022;36(4):818–826. doi: 10.1038/s41433-021-01535-9.