
ABSTRACT
Garcinia subelliptica Merr. (called “fukugi” in Japanese) is a dioecious tree species, which has traditionally been planted as windbreak forests in the coast of Ryukyu islands. Although effective afforestation and maintenance of G. subelliptica would greatly be assisted by early sex diagnosis and selection at the seedling stage, the genome and the sex-determination system of G. subelliptica are still poorly understood. In this study, we estimated the genome size of G. subelliptica to be approximately ~ 12 Gb for female and male, based on k-mer analysis. Furthermore, we developed a male-specific DNA marker for G. subelliptica, using the restriction site-associated DNA sequencing (RAD-seq) method. The 660 bp marker fragment, named FSDM (“fukugi” sex determination marker), was amplified in all males but not in the female G. subelliptica genome analyzed so far. In addition, the segregation ratio of the marker was 1:1 among the tested population. The DNA sequence of FSDM showed a significant similarity to the antifungal protein ginkbilobin-2 gene. The development of the sex-specific genetic marker of G. subelliptica may allow more effective conservation of traditional landscape and future design of villages around the northern part of the main island of Okinawa, a promising candidate site for World Natural Heritage.
Introduction
Garcinia subelliptica Merr. in the family Clusiaceae is native to the Ryukyu Islands of Japan, Taiwan and the Philippines (Robson Citation1996). Since this tree has excellent weather resistance, i.e. salty wind resistance and a heat relaxation effect, it has traditionally been planted to create windbreak forests in the Okinawa Prefecture (Itoh Citation2014). G. subelliptica is called “fukugi” in Japanese and has been a popular plant in Okinawa since the time of the Ryukyu Kingdom around 300 years ago. The northern part (Yambaru) of the main island of Okinawa is a candidate site for World Natural Heritage, and the village areas in Yambaru show over 50% tree coverage, where the half of the houses have G. subelliptica trees (Chen and Nakama Citation2010, Citation2011). This indicates that G. subelliptica is an integral plant species for the Yambaru villages and their ecosystem, and it seems to have profound impacts on the nature of Yambaru. The Garcinia genus comprises more than 250 dioecious species, from small shrubs to medium-sized trees that are common components of lowland tropical forests (Sweeney Citation2008). As a member of this genus, G. subelliptica is also a dioecious tree (Robson Citation1996; Taniguchi et al. Citation2008). The distinction between its male and female trees can only be discerned at the flowering stage, 10–12 years after germination. Mature female trees bear many fruits, which rot and give off a foul odor after they mature and fall, attracting pests (). Therefore, male trees that bear no fruit are preferred over female trees for planting around houses and along roads. Early diagnosis of sex at the seedling stage would greatly assist effective urban planning by desired roadside tree plantings, and also assist the maintenance of the windbreak G. subelliptica trees, the important landscape resource for the ecotourism in Yambaru. Registration to World Natural Heritage have a large impact on Japanese ecotourism, and would also raise public awareness of the importance of regional development and environmental protection (Song and Kuwahara Citation2016).
Figure 1. Flowers and fruits of the different sex types of G. subelliptica. (A) Male, (B) Female, (C) Hermaphrodite, (D) Fruits bearing in female tree, (E) Fallen fruit that produces an unpleasant smell

In several dioecious woody plants, sex determination DNA markers have been developed using various molecular techniques. Using randomly amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP) and polymorphic simple sequence repeat (SSR) methods, sex determination molecular markers have been developed for woody plants such as papaya (Carica papaya) (Urasaki et al. Citation2002), ginkgo (Ginkgo biloba) (Liao et al. Citation2009), date-plum (Diospyros lotus) (Akagi et al. Citation2014) and mulberry (Morus alba L.) (Atsumi et al. Citation2019). Furthermore, sex-specific molecular markers have proved to be very successful for inferring genetic sex determination mechanisms in dioecious plants.
Restriction site-associated DNA sequencing (RAD-seq) is a simple and rapid second-generation sequencing-based method with much higher throughput and lower cost than conventional methods (Baird et al. Citation2008). It is based on sequencing of short fragments from defined positions, flanking restriction enzyme recognition sites in the genome and counting their frequency. DNA polymorphisms among male or female individuals are represented by the presence or absence of these short sequences, i.e. tags. RAD-seq has been used to identify sex-specific markers in flowering plants, and all of these studies are based on experimental crosses and linkage map construction (Kafkas et al. Citation2015; Zhang et al. Citation2015). This approach is ideal as it can dissect the genetic architecture of sex determination in plants. RAD-Seq is often applied to artificially cross-population, but an artificial forest population was used in this study due to the long generation time of G. subelliptica. Therefore, the use of male and female individuals in natural populations serves as an alternative way to identify sex-specific markers by RAD-seq.
In this paper, we first estimated the genome size of G. subelliptica by using k-mer analysis based on short-read sequencing data. Next, we identified male-specific RAD-tag sequences of G. subelliptica using the RAD-seq method. The conversion of these RAD-tag sequences into a male-specific DNA marker allowed rapid sex identification in G. subelliptica. Moreover, the sex-determination system of G. subelliptica was inferred from the segregation ratio of a male-specific DNA marker using progeny obtained from 18 female trees.
Materials and methods
Plant materials and genomic DNA extraction
Plant materials used in the development of DNA markers are listed in . G. subelliptica populations were collected from four locations on the main island of Okinawa, Japan (Figure S1). We identified their sex, male or female, based on the morphology of flowers and fruiting phenotypes (). We used samples male and female 40 samples, respectively, from two of the locations (Yohena, Nago and Yamashiro, Itoman) for RAD-seq analysis. Genomic DNAs were extracted from young leaves (20 mg) of plant materials using the DNeasy Plant Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Nucleic acid concentration and quality were determined by NanoDrop 2000 c (Thermo Fisher Scientific, Waltham, MA, USA) and agarose gel electrophoresis.
Table 1. Individuals of G. subelliptica used in this study
Genome size estimation by k-mer analysis
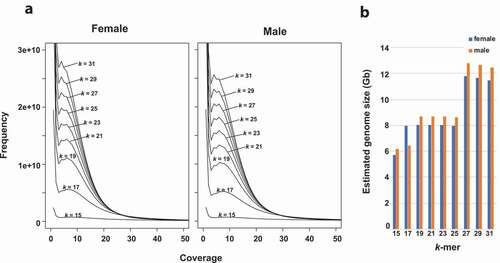
For the genome size estimation, we used a male and female plant DNA collected at Yohena, Nago (male) and Yamashiro, Itoman (female) (). The genome DNAs from female and male G. subelliptica were used for preparation of paired-end sequencing libraries, and were sequenced by Illumina NovaSeq6000 platforms at the OIST Sequencing Center (SQC) (Figure S2A). The 150 bp paired end reads (72 Gb for female and 78 Gb for male in total) were subjected to the k-mer frequency distribution analysis using Jellyfish v.2.0 (https://github.com/zippav/Jellyfish-2). Analysis parameters were set as -t 8 -C -s 5 G with -m 15 ~ 31, at steps of two, and the final results were plotted as frequency graphs. The genome size was calculated to be approximately 6 ~ 12 Gb for female and male, using the formula: Genome Size = k-mer Number/Peak Depth. This indicated that the sequenced short Illumina reads gave ~14 × coverage for the female and male genomes.
Preparation of RAD-seq, whole-genome sequencing (WGS) libraries
For RAD-seq analysis, bulked DNAs of 40 individuals from each male and female sample were used. One µg of pooled DNAs from male and female samples were digested with the six-base-cutting restriction enzyme PstI and the four-base cutter MseI (New England Biolabs, Beverly, MA, USA), which recognize CTGCAG and TTAA, respectively, and were purified using Agencourt AMPure XP (Beckman Coulter, Brea, CA, USA). Additionally, 1 µg of pooled DNAs from male and female samples were digested with MspI, which recognizes CCGG, and MseI (New England Biolabs), then were purified as above. Whole-genome sequencing (WGS) libraries of G. subelliptica for illumina sequencing were prepared from single male and female samples, in which DNA was fragmented to 350 bp with Covaris (Covaris, Woburn, MA, USA). To prepare samples for sequencing using HiSeq2500 platform (Illumina Inc.), double-digested DNAs and DNA fragmented to 350 bp were applied to the step “End Repair” in the TruSeq DNA LT Sample Prep Kit (Illumina Inc.) protocol. Six different indexed adapters were used for each sample. Adapter-ligated DNAs ranging in size from 300 bp to 1000 bp were checked using the Agilent Bioanalyzer 2100 (Agilent Technologies Inc, Santa Clara, CA, USA).
Sequencing and RAD-tag analysis
Four RAD-seq and two WGS libraries were pooled, respectively. The pooled sample was sequenced by HiSeq2500 (Illumina Inc.) with 150 bp paired-end flow cell. The resulting data (bcl files) were converted to fastq files for each of the six libraries by configureBclToFastq.pl. The MseIPstI and MseIMspI recognition sites used in the preparation of the RAD-seq library were observed at the 5´ ends in the RAD-tag sequences. Next, based on quality scores, 50 bases from 3´ ends in each read were trimmed. The 100 bp reads prepared from RAD-seq libraries were designated as RAD-tags and numbers of identical RAD-tags were counted in each sample. In the listed table, we excluded data showing less than 10 total RAD-tag counts for all samples. We extracted sex-specific RAD-tags by character-string comparison using Excel software from all RAD-tags data. RAD-tag sequences (100 bp) are recognized as different tags if even a single nucleotide is different. In exploration for male-specific RAD-tags, RAD-tags presented in only male bulk sample, and absented in female bulk sample were extracted. The female-specific RAD-tags were also explored in the same method. Thereafter, we performed BLASTN search using constructed male-specific contigs as the database, and male-specific RAD-tags extracted by Excel as the query. Contigs that completely contained male-specific RAD-tag sequences (100bp) were converted to DNA markers.
Construction of sex-specific contigs
RAD-seq reads were trimmed using Trimmomatic (https://github.com/timflutre/trimmomatic) with default parameters. The MEGAHIT assembler (https://github.com/voutcn/megahit) was used to assemble both RAD-seq and WGS data with default parameters. We then mapped male RAD-seq reads to female WGS assembly and vice versa by the program minimap2 (https://github.com/lh3/minimap2) with default parameters and extracted (via the in-house python script) male-specific and female-specific contigs using the mapped sequences and the assemblies (149,889 and 184,575 contigs for a male and female, respectively, Figure S2B). Next, to filter-out repetitive and complex sequences which were unsuitable for primer design, we performed sequence searches by BLASTX with the e-value 1e-6 as a threshold (Altschul et al. Citation1997) to the Uniport plant database, and extracted male- and female-specific contigs showing a match to known proteins. The sequences were used to design PCR primers.
Designing of DNA markers from male-specific RAD-tag sequence, and PCR conditions
In this study, to develop sex determination DNA markers, male-specific RAD-tags were analyzed, and female-specific RAD-tags were not analyzed. From the constructed male-specific contigs, primers were designed for randomly selected contigs completely containing male-specific tags and searched for sex-linked loci. The flanking region of a male-specific RAD-tag, named PMG1, was extracted from the contig sequences by BLAST (Altschul et al. Citation1997). Next, based on the extracted sequence (Contig k141_82390), forward primer PMG1F1 (5ʹ-ATTCGCTCAACATCACCTCC-3ʹ) and reverse primer PMG1R1 (5ʹ -TAACTGCGAGGTCTAGAAGT-3ʹ) on 5ʹ upstream and 3ʹ downstream of PMG1 were designed. Seven male and female DNA samples were used for PCR using the designed primer sets, and linkage to each sex was confirmed by agarose gel electrophoresis.
PCR amplification for DNA markers was performed in a 25-µL reaction mixture containing 0.25 µL KOD FX, 12.5 µL of 2× PCR Buffer for KOD FX, 5 µL of 2 mM dNTPs (TOYOBO, Japan), 40 pmol of each forward primer PMG1F1 and reverse primer PMG1R1, and 10 ng of genomic DNA. DNA was amplified in 30 cycles at 94°C for 10 sec, 55°C for 10 sec, 68°C for 1 min and final extension for 5 min at 68°C. The PCR amplicon was electrophoresed at 100 V for 25 minutes (0.5× Tris-Borate-EDTA buffer) on a 1.5% agarose gel containing ethidium bromide and detected under ultraviolet light (354 nm).
Sequencing and confirmation of a male-specific DNA marker fragment, FSDM
A sex-linked marker fragment, amplified by the primer set PMG1F1 and PMG1R1 from a male DNA, was named FSDM. Seven male PCR amplicons were directly sequenced by Applied Biosystems 3500 Genetic Analyzer using the BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific). To determine whether this DNA sequence is indeed male-specific, PMG1F2, PMG1F3, PMG1R2 and PMG1R3 oligo-nucleotide primers were designed inside FSDM. PCR was performed with nine combinations of the primers and positive control. A male and female individual were used as template DNA.
Validation of the DNA marker FSDM, and its segregation analysis in progeny
Plant materials () were used individually to examine the efficacy of FSDM for sex determination. PCR was performed using the primer set PMG1F1 and PMG1R1. After PCR and electrophoresis, the presence or absence of the amplification of FSDM band and the phenotype of G. subelliptica were validated. In addition, a total of 324 seedlings obtained from the progeny of 18 female trees at six locations () were used to investigate the segregation ratio of FSDM using the same method.
Results
Genome size estimation for G. subelliptica by k-mer analysis
To estimate the genome size of G. subelliptica, k-mer analysis was performed using the short read sequence data (The 150 bp paired end reads; 72 Gb for female and 78 Gb for male in total) obtained with Illumina NovaSeq6000 platform. The genome size estimation was performed with k values ranging from 15 to 31 at the step of two, showing the distribution curves with the peak depth of ~14 (). Based on the number of k-mers, the genome size of G. subelliptica was estimated to be approximately 6 ~ 12 Gb for female and male ().
Figure 2. The distribution of k-mers and the genome size estimation of G. subelliptica. (a) k-mer (k = 15 ~ 31) counts and their frequencies. (b) Estimated genome size based on the 15 ~ 31 k-mers
RAD-seq analysis
To develop DNA markers linked to sex, RAD-seq analysis was employed. Four RAD-seq libraries (male bulk; MseIPstI and MseIMspI, female bulk; MseIPstI and MseIMspI) from male and female bulked samples were sequenced by HiSeq2500 (Illumina Inc). The MseIPstI and MseIMspI recognition sites used in the preparation of the RAD-seq library were observed at the 5´ ends in the RAD-tag sequences. Next, based on quality scores, 50 bases from 3´ ends in each read were trimmed. The RAD-tag (100 bp) counts were calculated for each sample. In total, 325,505,402 and 119,662,827 RAD-tags were obtained from male bulk MseIPstI and MseIMspI, respectively. In the female bulk, 347,656,499 and 174,261,382 RAD-tags were obtained from MseIPstI and MseIMspI, respectively (Table S1). To develop DNA markers linked to the male locus, RAD-tags specific to male were extracted. The male-specific RAD-tag with a sum less than 10 were eliminated. In total, 1,836,590 and 1,564,522 unique male-specific RAD-tags were obtained for MseIPstI and MseIMspI, respectively. Male-specific RAD-tags were expected to be linked to the male locus.
Sex-specific contigs exploration, and conversion of RAD-tags into DNA markers
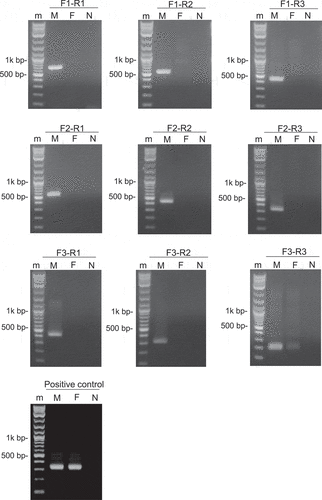
The summary of the male and female de novo assembly showed that N50 lengths were 1,146 and 1,144 bp, total lengths were 1,414,322,729 and 1,429,214,460 bp, number of contigs were 2,351,983 and 2,302,219, respectively. To develop sex-specific DNA markers, male RAD-seq reads were mapped to female WGS assemblies, and those that did not match were extracted as male-specific contigs (same method for female-specific contigs). In total, 149,889 male-specific and 184,575 female-specific contigs were obtained for male and female, respectively (Table S2). To explore flanking sequences of sex-specific RAD-tags, a BLASTN search was performed. As a result of male-specific contig exploration, 47 male-specific contigs containing male-specific RAD-tags were randomly selected and converted to DNA markers (Table S3). Among them, linkage of sex phenotypes was identified in only one contig (k141_82390) from 47 contigs and male-specific DNA markers could not be developed in the other 46 contigs. We succeeded in developing a male-specific DNA marker for the male-specific contig k141_82390 sequence (668 bp) including the RAD-tag (PMG1). The tag sequence (100 bp) count of the PMG1 was 28 in the male bulk sample, while for the female bulk sample it was 0 (Table S4). Next, a primer set PMG1F1 and PMG1R1 was designed at the 5ʹ end and 3ʹ end of a male-specific contig k141_82390 sequence (Figure S3), and PCR was performed. A single, distinct and highly reproducible band of around 600 to 700 bp was consistently amplified in male individuals, whereas no band was seen in female DNA samples (). This sex-linked fragment DNA marker was named FSDM (“fukugi” sex determination marker). Allele of FSDM was not found in the female contigs.
Figure 3. Results of PCR using primers: a primer set PMG1F1 and PMG1R1 designed from male-specific contig k141_82390. M: 1 kb Plus DNA Ladder (New England Biolabs), N: Negative control (H2O)

Sequencing of FSDM and confirmation of male-specific sequence
To confirm the DNA sequence of FSDM, sanger sequencing of seven male samples was performed using a primer set PMG1F1 and PMG1R1. The FSDM sequence was 660 bp in length with RAD-tag sequence (PMG1), that of seven male samples were the same. ClustalW analysis showed that the contig k141_82390 and FSDM were highly sequence homologous (Figure S4). BLASTX search for these sequences showed a significant similarity to the antifungal protein ginkbilobin-2 gene. To determine whether this DNA sequence is indeed male-specific, we designed PMG1F2, PMG1F3, PMG1R2 and PMG1R3 oligo-nucleotide primers inside FSDM and performed PCR with nine combinations (Table S5, Figure S4). The result was that FSDMs were amplified only from male DNA samples with the target size (Table S6), while FSDM amplification was not detected with female samples except a primer set PMG1F3 and PMG1R3 ().
Figure 4. Amplification pattern of FSDM locus in male and female individual. m: 1 kb Plus DNA Ladder (New England Biolabs); M: Male; F: Female; N: Negative control (H2O)
Validation of FSDM
To verify the effectiveness of FSDM to distinguish between female and male trees, DNAs of 101 males and 95 females () were individually tested by PCR with PMG1F1 and PMG1R1 primers. By comparing the male/female phenotypes and PCR results, we confirmed that FSDM was amplified from all males and none from females ().
Table 2. Sex identification using FSDM
Segregation ratio of FSDM
We further investigated the segregation of the presence or absence of FSDM amplification using 324 seedlings (), showing that 166 seedlings were FSDM positive and 158 seedlings were FSDM negative. The observed ratio of presence to absence was not significantly different from 1:1 in total (P > 0.1) ().
Table 3. Segregation of FSDM in progeny of 18 female trees
Discussion
RAD-seq analysis can generate a great number of unlinked polymorphic markers for non-model organisms while substantially reducing labor and sequencing costs than conventional methods (Zhou et al. Citation2018). So far, there is a report that the chromosome number of G. subelliptica is 2 n = 32 (Nakazato Citation1994), but little is known about the genome size and the sequence of G. subelliptica. In this study, we estimated the genome size for both female and male G. subelliptica using short-read sequences, and also used the RAD-seq method to develop a sex-identifying DNA marker for G. subelliptica.
In the genus Garcinia, only the genome size of mangosteen (G. mangostana) has been reported. According to Midin et al. (Citation2018), the genome size of mangosteen was estimated to be approximately 5.92 Gb using k-mer analysis. In this study, the genome size of G. subelliptica was estimated to be approximately ~12 Gb for female and male, which is relatively larger than mangosteen and other plant species. However, further analyses would be needed to determine the genome size more accurately.
By RAD-seq analysis, we obtained 320 million and 340 million RAD-tags digested with MseI and PstI for male and female genomes, respectively. The unique number of RAD-tags that exist only in male-specific samples was 1,836,590, which can be further used for the development of DNA markers linked to male-specific genomic regions in future studies. The RAD-seq method has been used in the development of DNA markers for crop plants such as leaf margin phenotype-specific markers of pineapple (Ananas comosus) (Urasaki et al. Citation2015) and DNA markers associated with gynoecy in bitter gourd (Momordica charantia) (Matsumura et al. Citation2014) without genome information. Because woody plants have long generation times compared with other flowering plants, genetic studies of sex determination for woody plants by experimental crossing are generally impractical. However, the RAD-seq method can directly sample bulked males and females from a natural population, thereby reducing the cost and labor compared to conventional methods, without using experimental crossing. It has become possible to develop sex-specific DNA markers for pistachio (Pistacia vera) (Kafkas et al. Citation2015), kiwi (Actinidia chinensis) (Zhang et al. Citation2015) and mangrove (Excoecaria agallocha) (Zhou et al. Citation2018) using the RAD-seq method. In this study, by applying the RAD-seq method, we were able to develop a male-specific DNA marker from G. subelliptica, a non-model woody plant.
We demonstrated that the male-specific DNA marker, named FSDM, was consistent with the tree sex phenotypes and the genome DNAs for 101 males and 95 females collected from four different locations on the island of Okinawa. Importantly, the FSDM amplification by PCR was independent of sampling location and tree age, suggesting that this DNA marker is useful for early sex diagnosis of G. subelliptica. The DNA sequence of FSDM was found only in male trees, and exhibited sequences showed significant similarity to the antifungal protein ginkbilobin-2 (GNK2) gene. Interestingly, GNK2 encodes an antifungal protein in ginkgo (G. biloba), a dioecy tree species like G. subelliptica (Sawano et al. Citation2007). One in a total of 196 trees was identified as a “mix” tree consisting of male and hermaphrodite flowers (, ). As a result of PCR, FSDM was amplified from the “mix” tree. Regarding the “mix” phenotype tree, Taniguchi et al. (Citation2008) reported one “mix” out of 46 trees. Nakazato et al. (Citation1992) also reports 51 “mix” trees out of 802 trees. Previously, Lin et al. (Citation2016) reported that papaya shows the male-to-hermaphrodite sex reversal at low temperatures. This is caused by silencing of the gynoecium suppression function on the sex determination pathway through epigenetic modification. The sex reversal in the “mix” trees of G. subelliptica may also be due to environmental factors as papaya, while the frequency of the reversal phenotype is very low. In addition, since the number of fruits on the “mix” tree is much fewer than that of the female trees, there is no significant effect on pests attracted by the fruits.
Regarding the sex ratio of G. subelliptica, Taniguchi et al. (Citation2008) reported that there were more male trees than female trees at two locations, although their report examined a small number of samples (n = 32 and 46). Nakazato et al. (Citation1992) also reported that there were slightly more male than female trees in a large-scale investigation (n = 802). Based on their study, we expected a lower female ratio of G. subelliptica in 324 seedlings obtained from 18 female trees. However, the ratio of FSDM amplification was not significantly different from the expected segregation ratio 1:1, indicating that the sex ratio of G. subelliptica is close to 1:1. The sex ratio surveys of G. subelliptica trees by Taniguchi et al. (Citation2008) and Nakazato et al. (Citation1992) were carried out by mature trees in an artificial forest and homestead woodland. The existence of slightly more male trees than female trees may come from the artificial removement of female trees in the course of forestation. Genetic sex determination systems in dioecious trees are known as XY and ZW types (Kersten et al. Citation2017). These results may suggest that FSDM is located on or closely linked to the male-specific region of the Y chromosome (MSY) in G. subelliptica. On the other hand, a large number of female-specific contigs were not analyzed in this study, and therefore whether there is a X chromosome in female genome of G. subelliptica is currently unclear. Further studies would reveal the sex-determination system of G. subelliptica.
In this report, we showed that FSDM converted from a male-specific RAD-tag can be used to determine the sex of a G. subelliptica tree at an early developmental stage. The sex was correctly predicted with this marker in each of the 196 G. subelliptica trees tested. The application of the FSDM for sex identification of G. subelliptica at the seedling stage will enormously facilitate the cultivation and greening of urban space by desired G. subelliptica trees with less time, space and labor cost. For example, many G. subelliptica trees are planted in the villages around the northern part of the main island of Okinawa, which have been contributed to the maintenance of the protective Yambaru area. However, a progression of depopulation in the area in recent years make it difficult to maintain the landscape of Yambaru villages, which may become an issue for its registration to the World Heritage, and also for the future ecotourism. As the result of this study, we are now able to select and plant male plants as homestead woodland and roadside in the village, and female plants as windbreak forest along the coast, which may eventually support the protection and conservation of the landscape in Yambaru area suitable for the World Heritage.
Data archiving statement
Raw sequencing data of the Illumina reads have been deposited in the DDBJ Sequence Read Archive (DRA) under accession number DRA010543.
Acknowledgments
This study was supported by the Okinawa Prefecture Industrial Research Promotion Project.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Akagi T, Kajita K, Kibe T, Morimura H, Tsujimoto T, Nishiyama S, Kawai T, Yamane H, Tao R. 2014. Development of molecular markers associated with sexuality in Diospyros lotus L. and their application in D. kaki Thunb. J Japan Soc Hort Sci. 83(3):214–221. doi:10.2503/jjshs1.CH-109.
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25(17):3389–3402. doi:10.1093/nar/25.17.3389.
- Atsumi R, Nishihara R, Tarora K, Urasaki N, Matsumura H. 2019. Identification of dominant genetic markers relevant to male sex determination in mulberry (Morus alba L.). Euphytica. 215(11): 187.
- Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA. 2008. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PloS One. 3(10):e3376. doi:10.1371/journal.pone.0003376.
- Chen B, Nakama Y. 2010. A study on village forest landscape in small island topography in Okinawa, Japan. Urban Forestry & Urban Greening. 9(2):139–148. doi:10.1016/j.ufug.2009.12.004.
- Chen B, Nakama Y. 2011. Distribution of Fukugi (Garcinia subelliptica) trees as landscaping trees in traditional villages in Ryukyu Islands in Japan. Pac Agric Nat Resour. 3:14–22.
- Itoh S. 2014. Decline and blight of Garcinia subelliptica at Nakadomari, Onna village in mid part of Okinawa island. Kyushu J. For Res. 67: 25–28. in Japanese.
- Kafkas S, Khodaeiaminjan M, Güney M, Kafkas E. 2015. Identification of sex-linked SNP markers using RAD sequencing suggests ZW/ZZ sex determination in Pistacia vera L. BMC Genomics. 16(1):98. doi:10.1186/s12864-015-1326-6.
- Kersten B, Pakull B, Fladung M. 2017. Genomics of sex determination in dioecious trees and woody plants. Trees. 31(4):1113–1125. doi:10.1007/s00468-017-1525-7.
- Liao L, Liu J, Dai Y, Li Q, Xie M, Chen Q, Huaqun Y, Guanzhou Q, Liu X. 2009. Development and application of SCAR markers for sex identification in the dioecious species Ginkgo biloba L. Euphytica. 169(1):49–55. doi:10.1007/s10681-009-9913-8.
- Lin H, Liao Z, Zhang L, Yu Q. 2016. Transcriptome analysis of the male-to-hermaphrodite sex reversal induced by low temperature in papaya. Tree Genet Genomes. 12(5):94. doi:10.1007/s11295-016-1055-2.
- Matsumura H, Miyagi N, Taniai N, Fukushima M, Tarora K, Shudo A, Urasaki N. 2014. Mapping of the gynoecy in bitter gourd (Momordica charantia) using RAD-seq analysis. PloS One. 9(1):e87138. doi:10.1371/journal.pone.0087138.
- Midin MR, Nordin MS, Madon M, Saleh MN, Goh HH, Mohd NN. 2018. Determination of the chromosome number and genome size of Garcinia mangostana L. via cytogenetics, flow cytometry and k-mer analyses. Caryologia. 71(1):35–44. doi:10.1080/00087114.2017.1403762.
- Nakazato N. 1994. Studies on the sexuality of Garcinia subelliptica Merr. (I) layering and chromosome number of seedling. Transactions of the Japanese Forestry Society. 105: 317–318. in Japanese.
- Nakazato N, Nagano K, Toda Y. 1992. Study on the sex of Garcinia subelliptica Merr. flower and sex ratio. Kyushu J For Res. 45: 35–36. in Japanese.
- Robson NKB. 1996. Guttiferae in flora of taiwan. Vol. 2, 2nd ed. ed. Taipei (Taiwan): Editorial Committee of the Flora of Taiwan. p. 694–714.
- Sawano Y, Miyakawa T, Yamazaki H, Tanokura M, Hatano KI. 2007. Purification, characterization, and molecular gene cloning of an antifungal protein from Ginkgo biloba seeds. Biol Chem. 388(3):273–280. doi:10.1515/BC.2007.030.
- Song D, Kuwahara S. 2016. Ecotourism and world natural heritage: its influence on islands in Japan. J Mar Island Cult. 5(1):36–46. doi:10.1016/j.imic.2016.05.006.
- Sweeney PW. 2008. Phylogeny and floral diversity in the genus Garcinia (Clusiaceae) and relatives. Int J Plant Sci. 9(2):1288–1303. doi:10.1086/591990.
- Taniguchi S, Nishihara F, Nakasuga T. 2008. Studies of gender in flower production, survival and maturity process of fruit in dioecious Garcinia subelliptica Merr. trees. Kyushu J. For Res. 61: 21–25. in Japanese.
- Urasaki N, Goeku S, Kaneshima R, Takamine T, Tarora K, Takeuchi M, Moromizato C, Yonamine K, Hosaka F, Terakami S, et al. 2015. Leaf margin phenotype-specific restriction-site-associated DNA-derived markers for pineapple (Ananas comosus L.). Breed Sci. 65(3):276–284. doi:10.1270/jsbbs.65.276.
- Urasaki N, Tokumoto M, Tarora K, Ban Y, Kayano T, Tanaka H, Oku H, Chinen I, Terauchi R. 2002. A male and hermaphrodite specific RAPD marker for papaya (Carica papaya L.). Theor Appl Genet. 104(2):281–285. doi:10.1007/s001220100693.
- Zhang Q, Liu C, Liu Y, VanBuren R, Yao X, Zhong C, Huang H. 2015. High-density interspecific genetic maps of kiwifruit and the identification of sex-specific markers. DNA Research. 22(5):367–375. doi:10.1093/dnares/dsv019.
- Zhou Y, Wu W, Ning Z, Zhou R. 2018. Identification and characterization of sex-specific markers in the milky mangrove Excoecaria agallocha using double digest restriction site-associated DNA sequencing. Aquat Bot. 5:54–60. doi:10.1016/j.aquabot.2017.11.004.