
Abstract
From a clinical perspective, there is a need for a reliable and comprehensive list of diseases causing AA amyloidosis. This list could guide clinicians in the evaluation of patients with AA amyloidosis in whom an obvious cause is lacking. In this systematic review, a PubMed, Embase and Web of Science literature search were performed on causes of AA amyloidosis published in the last four decades. Initially, 4066 unique titles were identified, but only 795 full-text articles and letters were finally selected for analysis. Titles were excluded because of non-AA type of amyloidosis, language, no full-text publication or irrelevance. Hundred and fifty diseases were initially reported to be associated with the development of AA amyloidosis. The presence of AA amyloid was proven in 208 articles (26% of all) of which 140 (67%) showed a strong association with an underlying disease process. Disease associations were categorized and 48 were listed as strong, 19 as weak, 23 as unclear, and 60 as unlikely. Most newly described diseases are not really unexpected because they often cause longstanding inflammation. Based on the spectrum of identified causes, a pragmatic diagnostic approach is proposed for the AA amyloidosis patient in whom an obvious underlying disease is lacking.
Introduction
Systemic AA amyloidosis, previously known as secondary or reactive amyloidosis, is a long-recognized severe complication of some chronic inflammatory diseases. In AA amyloidosis organ damage results from the extracellular deposition of the soluble acute-phase reactant serum amyloid A (SAA) protein as insoluble amyloid fibrils. A sustained high concentration of SAA produced by the liver during a chronic inflammatory state is the prerequisite for developing AA amyloidosis [Citation1].
In the past, pathologists observed organs heavily affected by amyloid in post-mortem studies of infectious diseases such as tuberculosis (TBC), syphilis and osteomyelitis [Citation2]. Later also myeloma was found to be associated with amyloid deposition [Citation3]. The isolation of amyloid fibrils by extraction with distilled water in 1968 made it possible to chemically analyse amyloid [Citation4]. In myeloma-associated amyloid, the fibrils were derived from immunoglobulin light chains; therefore this type was later called AL type amyloid [Citation5]. In inflammation-associated amyloid, the fibrils were derived from an unknown protein that was later designated AA type [Citation6]. An acute phase reactant in serum that was immunochemically related to AA amyloid led to the identification of SAA [Citation7]. Several inflammatory diseases and other diseases leading to longstanding SAA production (e.g. rheumatoid arthritis) were associated with chemically and immunochemically defined amyloid of the AA type [Citation6,Citation8–14].
The introduction of the rectum biopsy combined with the Congo red stain, after 1950, enabled clinicians to detect amyloid during life [Citation15]. The detection with the electron microscope of fine, non-branching, rigid fibrils of indeterminate length and about 5–14 nm wide became another diagnostic characteristic of amyloid [Citation16]. The sensitivity of the Congo red stain after pre-treatment with potassium permanganate (KMnO4) was the first test that helped to differentiate AA amyloid from other types of amyloid [Citation17], being useful in clinical practice at that time [Citation18]. Later, immunohistochemistry became the method of choice to identify different types of amyloid in clinical practice [Citation19]. However, immunohistochemistry appears not to be fully reliable in all cases either [Citation20]. The latest identification improvement was proteomics, applied to micrograms of tissue and resulting in chemical determination of the type of amyloid [Citation21]. The biopsy results enabled many authors to describe series and individual patients with AA amyloidosis, thereby identifying various underlying diseases over the last 40–50 years.
A causal relation between a disease and AA amyloidosis can be proven in animal studies in reproducible settings, such as subcutaneous inflammation in mice [Citation22] or hamsters [Citation23], alveolar hydatid disease in mice [Citation24], Opisthorchis viverrini infection in hamsters [Citation25] and Enterococcus faecalis infection in chickens [Citation26]. Such a direct causal relation cannot be established in human beings. From a clinical perspective causality can be assumed because of overwhelming evidence in the literature on the development of AA amyloidosis in some inflammatory diseases, such as rheumatoid arthritis. However, the question of coincidence rises in single cases of rare inflammatory diseases that have been found together with AA amyloidosis. Causality becomes more plausible if at least two different cases with AA amyloidosis are both found to be suffering from the same rare inflammatory disease. Causality is also highly likely in cases with AA amyloidosis where organ damage reverses dramatically and continuously for years after successfully treating the underlying disease.
Lists of diseases causing AA amyloidosis have been published [Citation1,Citation27–29]. From a clinical perspective, there is a need for an updated, reliable and comprehensive list. This list could guide clinicians in the evaluation of patients with AA amyloidosis in whom an obvious cause is lacking. Identification of the underlying cause provides opportunities for targeted therapy of the inflammatory process, thereby significantly improving the prognosis of these patients [Citation30]. There is also a need for a systematic diagnostic approach that can be used in any patient with AA amyloidosis in whom an obvious cause is lacking.
The aim of this systematic review is to list all diseases published in the last four decades that are reported as causally related to AA amyloidosis. The strength of the evidence of AA amyloid and the association with the underlying disease will be studied. This list of diseases will be used to propose a pragmatic diagnostic approach of patients newly diagnosed with AA amyloidosis. Furthermore, the intriguing disease entity of idiopathic AA amyloidosis will be discussed.
Methods
Search strategy
Inclusion criteria were case reports or series of patients diagnosed with systemic AA amyloidosis. Exclusion criteria were (1) case reports or series describing non-AA type of amyloidosis or localized amyloidosis, (2) articles in languages other than English, French, German or Dutch and (3) no full-text publication. PubMed, Embase and Web of Science were used. Search queries used are presented in the Supplementary file. References of the included articles were screened to find more articles. For all diseases that only showed a weak, unclear or unlikely association (as described below) a second search was performed in PubMed with the terms: “specific illness” AND “amyloidosis”.
Diagnosis of AA amyloid
The diagnosis of AA amyloid was classified as proven, likely, possible or problematic. Proven diagnosis: amyloid deposits had to be characterized histochemically by positive staining with Congo red, which also showed typical anomalous colours under polarized light, or the amyloid deposits had to be characterized by electron microscopy [Citation31]. AA type had to be identified by immunohistochemistry or by proteomics [Citation21,Citation31,Citation32]. There could not be any suspicion or evidence of other types of amyloid. Likely diagnosis: amyloid deposits had to be characterized histochemically by positive staining with Congo red, which also showed typical anomalous colours under polarized light or the amyloid deposits had to be characterized by electron microscopy [Citation31]. Based on clinical findings and the distribution pattern of amyloid, AA amyloid is the most likely diagnosis, but identification of AA type using immunohistochemistry or proteomics is absent. Possible diagnosis: amyloid deposits had to be characterized histochemically by positive staining with any of the usual amyloid stains, including immunohistochemistry. There could not be evidence of non-AA amyloid. Problematic diagnosis: no report on any results of one of the usual amyloid stains. Or in case there is evidence of a non-AA type of amyloid.
Strength of the association of an underlying disease with AA amyloidosis
The strength of the association between the underlying disease and development of AA amyloidosis was defined as strong, weak, unclear or unlikely. Strong association: two or more publications of different groups that describe the association between one particular disease and the development of AA amyloidosis, in which the diagnosis of AA amyloid is proven (as described above). Weak association: two or more publications that describe the association between one particular disease and the development of AA amyloidosis, in which the diagnosis of AA amyloid is at least likely (as described above). Unclear association: two or more publications that describe the association between only one particular disease and the development of AA amyloidosis, in which the diagnosis of AA amyloid is at least possible (as described above). Further supportive evidence is lacking. Unlikely association: a publication in which an association had been assumed, but there was more than one underlying disease, or other diseases seem to have been the more likely cause of AA amyloidosis, or in which a diagnosis of AA amyloid is in retrospect deemed unlikely or not supported at all. Or only one single publication of a disease with patients having proven, likely, or possible AA amyloid: coincidence cannot be excluded in such a situation.
Data collection and analysis
Three authors independently assessed articles for inclusion. Articles included in the final table were reviewed by all three authors. In case of disagreement, diagnosis of AA amyloid and strength of associations were discussed until consensus.
Results
Literature search
Titles found: PubMed: total number 2596, Embase: total number 3359, Web of Science (core collection): total number 2552. Total titles PubMed, Embase and Web of Science: 8507, search date 05-06-2018.
Selection of the publications
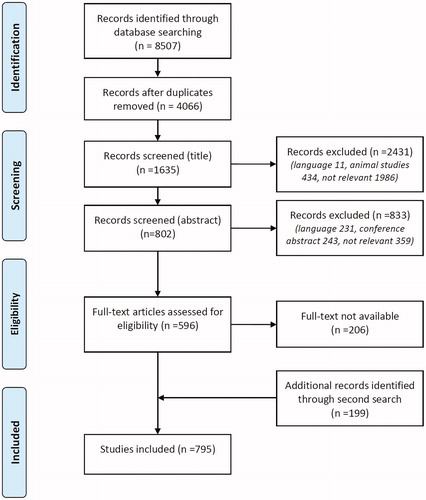
First 4441 duplicates were removed. Of the remaining 4066 unique titles 2431 were excluded because the articles were written in languages other than English, French, German or Dutch (11), concerned animal studies (434), or were not relevant (1986), leaving 1635 titles. After reading the abstract 833 publications were excluded because of language (231), presentation/conference abstract only (243), or deemed not relevant (359), leaving 802 publications. Of 206 publications reprints were not readily available and not deemed necessary for the analysis (since a strong association with the disease could already be established with the directly available reprints, e.g. rheumatoid arthritis), leaving 596 publications. A second search based on references of the included publications and a new PubMed search looking for the combination of amyloidosis and the specific disease yielded another 199 articles, resulting in a final number of 795 full-text publications available for analysis ().
Figure 1. Systematic view of database search and case selection.
Overview of the study group
Hundred and fifty diseases were initially reported to be associated with the development of AA amyloidosis. Ninety-four of the 795 publications presented information concerning more than one disease. Electron microscopy was described in 107 publications (13% of all). Congo red-stained tissue was described in 560 (70%) publications of which birefringence was described in 310 (55%), the potassium permanganate test in 140 (25%), and anti-AA immunohistochemistry in 310 (55%). Amyloid A was proven in 207 (26% of all) publications of which 140 (68%) were strongly associated with an underlying disease process. The quality of the immunohistochemistry was seldom checked by using a positive and negative control and criteria of the underlying inflammatory disease were not described consistently among the various diseases.
Disease associations
lists 48 diseases that were found to be strongly associated with the development of AA amyloidosis and lists 19 diseases that were found to be weakly associated. All diseases have been categorized under the following seven headings: chronic infection, chronic inflammation, hereditary, haematologic disease, tumour, miscellaneous and idiopathic. Twenty-three diseases that were unclearly associated with the development of AA amyloidosis have been listed in . A list of 60 diseases in which the association with development of AA amyloidosis was unlikely is presented in . In the Supplementary file, two representative references are presented for each disease together with more detailed information about the associations. The numbers of strongly associated references and of all retrieved references are presented in Supplementary Tables 1–4. Presence or absence of underlying disease criteria, of the acute phase response and of the clinical picture of AA amyloidosis are also presented in the Supplementary tables.
Table 1. Strong associations with AA amyloidosis.
Table 2. Weak associations with AA amyloidosis.
Table 3. Unclear associations with AA amyloidosis.
Table 4. Unlikely associations with AA amyloidosis.
Discussion
Sixty-seven of the 150 investigated diseases (45%) were strongly () or weakly () associated with AA amyloidosis whereas there was no clear association in 23 diseases (15%) (). It is striking that the use of Congo red stain to make the diagnosis was reported in only 70% of the publications and it is even disappointing that the additional use of polarized light to actually prove the presence of amyloid was only reported in about half of these publications. Although immunohistochemistry with anti-AA was used in 55% of the Congo red-stained tissues, it was also notable that the obsolete potassium permanganate test was used in 25% of these cases and reported to be diagnostic for AA amyloid. It was not possible to assess the quality of the anti-AA immunohistochemistry because the use of positive and negative control tissues was only seldom reported. The documented criteria of the underlying disease varied considerably among the diseases and were frequently not reported at all. Exclusion of other possible underlying diseases was also variably reported and not possible to evaluate because of the huge difference in diagnostic possibilities within the studied decennia.
Assessment of associations compared to previous lists
Most (43 out of 49 [Citation1], 35 out of 41 [Citation28] and 44 out of 58 [Citation29]) of the associated diseases listed earlier in the literature appear to be strongly or weakly associated ( and ). Fifteen diseases have now been assessed differently. Polyarteritis nodosa (categorized as vasculitis, not further specified), sickle cell anaemia, gastrointestinal stromal tumour (GIST) and human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDS) are presented as unclearly associated in . HIV/AIDS as direct cause because of viral infection (HIV) was deemed unclearly associated. HIV/AIDS as indirect cause because of immunodeficiency (AIDS) was also unclearly associated because it was generally found in combination with skin popping, intravenous drug abuse, or other earlier or concurrent infections instead of a subsequent infection. Jejuno-ileal bypass was deemed unlikely in because morbid adiposity is the logical pre-existing underlying condition. Schnitzler’s syndrome, primary ciliary dyskinesia (PCD), antineutrophil cytoplasmic antibody-associated (ANCA)-vasculitis and hairy cell leukaemia were also deemed unlikely since there was only one well-described case for these diseases. For both hyper immunoglobulin M (IgM) syndrome and SAPHO (synovitis, acne, pustulosis, hyperostosis and osteitis) syndrome three case reports were found, in none of which amyloid was characterized. Subacute bacterial endocarditis, inflammatory abdominal aortic aneurysm and myxoma were only once reported in the last decennia, whereas references concerning carcinoid tumour were not found.
New associations
Eighteen new associations have been added to and (marked with an asterisk). Many newly described diseases are not unexpected because they often cause longstanding inflammation, such as hidradenitis suppurativa, acne conglobata, xanthogranulomatous pyelonephritis and several other infectious diseases (non-tuberculosis mycobacterium, abdominal infection/abscess, malaria, schistosomiasis, filariasis and aspergillosis). A remarkable cause is alkaptonuria [Citation33]. This association was described several times by just one group and would need confirmation by another group. It is possible that the disease causes inflammation by ochronotic arthritis of the spine and large joints associated with an elevated ESR in 20% of the patients [Citation34]. However, a clear clinical picture of systemic AA amyloidosis with proteinuria, loss of renal function or other systemic symptoms was not reported. Another less obvious cause is glycogen storage disease (GSD). Increased SAA production in GSD can be caused by the development of hepatocellular adenomas [Citation35], observed in 57% of the patients with GSD type 1 [Citation36,Citation37], or due to recurrent infections[Citation38], caused by neutropenia and neutrophil dysfunction. SAA production in hepatocytes is stimulated by the production of high amounts of tumour necrosis factor-alpha (TNF-α) [Citation39]. In inflammatory hepatocellular adenomas, SAA is overexpressed by neoplastic hepatocytes [Citation40]. Coeliac disease may cause AA amyloidosis since T-cell driven inflammation by gluten ingestion results in the release of pro-inflammatory cytokines, although elevated CRP or SAA levels have not been reported [Citation41].
Rheumatic diseases and viral infections
Strong disease associations were established in mixed connective tissue disease (MCTD), systemic lupus erythematosus (SLE) and hepatitis B/C. This was not directly expected as systemic connective tissue diseases and viral infections are usually accompanied by lower SAA levels than other rheumatic diseases, like rheumatoid arthritis (RA), and bacterial infections. Where polymyalgia rheumatica (PMR) was often followed by giant cell arteritis (GCA), the combination was more strongly associated with AA amyloidosis than PMR alone.
Associated tumours
The list of tumours in and has become more diverse, however, most tumours are only weakly associated with AA amyloidosis and would need further confirmation. Non-Hodgkin’s lymphoma is strongly associated with AA amyloidosis, but, like Waldenström’s disease, its close association with monoclonal immunoglobulins and light chains requires that AL amyloid always must be excluded without any doubt. Proving the actual presence of AA amyloidosis in Waldenström’s disease or lymphomas means that treatment of these underlying diseases with even high-dose chemotherapy may be indicated to effectively treat AA amyloidosis in these patients [Citation42]. In cases describing oesophageal cancer and colorectal carcinoma, a clinical picture of systemic AA amyloidosis is missing and for pancreatic carcinoma the described clinical picture raises even doubt concerning the diagnosis. Immunohistochemistry is missing in cases describing bladder, colorectal and giant basal cell carcinoma (GBCC). However, longstanding skin ulceration in GBCC fits seamlessly. Overall, it is thought that amyloid development in neoplastic diseases is caused by tumour-induced inflammation. There is often a granulomatous inflammatory component in malignant disease [Citation43]. Besides, it has become evident that an inflammatory microenvironment is an essential component for tumour development [Citation44]. Twenty per cent of cancers are linked to chronic infections (e.g. Hodgkin’s disease and large B cell lymphoma: Epstein–Barr virus; colon and bladder carcinoma: Schistosoma or Bacteroides species), 30% to tobacco smoking and inhaled pollutants (e.g. lung carcinoma and mesothelioma) and 35% to dietary factors (with 20% of cancer burden related to obesity) [Citation45]. Therefore, one can speculate that this inflammatory microenvironment observed during tumour development also contributes to the development of AA amyloidosis in a similar way as to that observed in obesity [Citation46].
Overlapping associations
Several diseases associated with AA amyloidosis overlap, as seen in Rosai–Dorfman histiocytosis and immunoglobulin G subtype 4 (IgG4)-related diseases, making it difficult to establish the association with AA amyloidosis on its own [Citation47]. Such a relation is also described in paraganglioma and GIST, which are linked by a common germline mutation in the succinate dehydrogenase (SHD)-gene known as the Carney–Stratakis syndrome [Citation48]. However, this relation has not been described yet in cases with AA amyloidosis. Paragangliomas sometimes secrete inflammatory cytokines, such as interleukin-6 (IL-6) [Citation49], which also has been described in GIST [Citation50]. However, the association of GIST with AA amyloidosis remains doubtful. In one case, the clinical picture suggested AA amyloidosis [Citation50], however, in two other cases the clinical picture was either not described [Citation51] or there were other more plausible disease associations (morbid obesity and gout) [Citation52]. What we did not investigate, but what the above-mentioned cases do illustrate is that AA amyloidosis can be caused by more than one disease. This makes a proper clinical diagnosis very important, but also the recognition of all concurrent diseases that increase the SAA serum levels in the patient involved.
Infectious causes
Amyloidosis is more commonly caused by infections than currently described. Modern successful treatment of infections prevents a longstanding inflammatory course. In older literature, we could find many cases of infections followed by amyloidosis as cause of death. In 1973, a series of 112 patients with visceral actinomycosis was described of whom 14 had AA amyloidosis [Citation53]. Another other example is syphilis, originally known as the third most common cause of AA amyloidosis after tuberculosis and osteomyelitis [Citation54]. Nowadays, the disease has vanished almost completely, leaving only an unclear association. Periodontitis on the other hand, may be a more frequent cause than described. In cases described by Cengiz (Suppl. ref. [Citation35]) longstanding (decennia) and severe destructive inflammation of the dental region may be a plausible underlying cause. Diseases such as filariasis and echinococcosis are rare in the Western world and therefore may be underestimated.
Some diseases are associated with the development of AA amyloidosis because patients become prone to (recurrent) infections. This is the case in patients with immunodeficiencies (e.g. common variable immune deficiency, hypogammaglobulinemia), or in a disease such as cystic fibrosis. In paraplegia, the development of (chronic) decubitis and urinary tract infections are the underlying causes of sustained SAA production. Recurrent pulmonary infections may cause bronchiectasis which itself is strongly associated with AA amyloidosis.
Unclearly associated conditions with potential for strength
Not only infections, but also other diseases listed as unclearly associated in are potential candidates for a strong association. For example, cases describing sickle cell anaemia were convincing, but were not listed as weak or strong association because birefringence was not reported. A similar situation is observed in systemic sclerosis, primary sclerosing cholangitis, autosomal dominant polycystic kidney disease, pulmonary alveolar proteinosis, chronic granulomatous disease, and Langerhans’ cell histiocytosis.
Unlikely associations
Thirty-eight of the 60 diseases listed in were reported only once. Schnitzler’s syndrome, ANCA-associated vasculitis and PCD are diseases that might be more strongly associated, but lack a second case with well-described histology. Although other unlikely associated diseases were described more often, descriptions were too vague or lacking (e.g. Leishmaniasis, SAPHO syndrome, myxoma, inflammatory pseudotumor and tumoral calcinosis). Inflammatory diseases such as septic arthritis and reactive arthritis were more likely to be related to underlying RA and TBC, or to spondyloarthropathy, obesity or inflammatory bowel disease, respectively. Pleura empyema and aneurysm were related to pre-existing bronchiectasis, endocarditis or TBC, and cases with myositis were related to underlying neoplastic disorders. In Sjögren’s syndrome, multiple myeloma and MGUS amyloidosis of AL type was more plausible. In patients on haemodialysis β2-microglobulin-associated (Aβ2M) amyloidosis and in the patient with multifocal motor neuropathy transthyretin-associated (ATTR) type amyloidosis were more plausible than AA amyloidosis. Finally, chronic lymphatic leukaemia and ureterosigmoidostomy were excluded from this review, since full-text papers could not be retrieved [Citation55,Citation56].
Development of AA amyloidosis
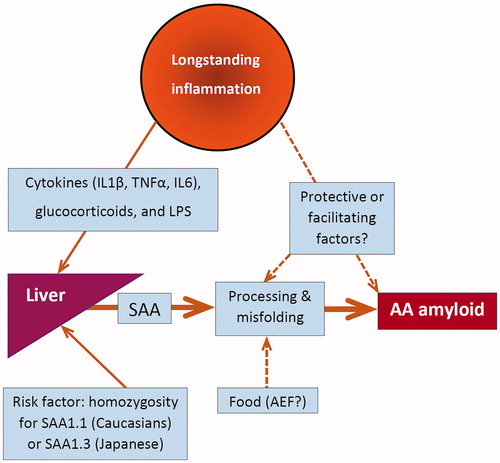
The production of SAA is regulated by cytokines such as interleukin-1 (IL1), interleukin-6 (IL6) and TNF, but also by glucocorticoids and lipopolysaccharide (LPS) [Citation57]. The liver is the main site of SAA production, but monocyte-derived macrophages also can produce SAA [Citation58]. Although chronically elevated SAA serum levels are a prerequisite, they are not sufficient alone to develop AA amyloidosis (). Even in longstanding and severe inflammatory rheumatic diseases no more than 10–30% of the patients develop amyloidosis [Citation59,Citation60]. The question arises whether there are protective factors in these patients with chronic inflammation but who do not develop amyloidosis or facilitating factors other than elevated SAA in patients who do develop amyloidosis. Fibril-derived amyloid-enhancing factor (AEF) appeared to be such a facilitating factor in animal studies and AA amyloidosis appeared to be transmissible by oral ingestion of food containing AA-amyloid in a mouse model. These observations suggest that exposure to exogenous substances with AEF activity might facilitate the development of AA amyloidosis in susceptible patients [Citation61]. The SAA1 genotype is another factor probably affecting amyloidogenesis. In Caucasians, the SAA1.1 allele and in Japanese homozygosity for the SAA1.3 allele was associated with AA amyloidosis in particular patient groups [Citation59,Citation62]. It is intriguing whether the specific pathogenic mechanism underlying the inflammatory process plays a role in amyloidogenesis. In this respect, it is notable that granulomatous inflammation is frequently present in diseases associated with AA amyloidosis (e.g. TBC, leprosy, rheumatoid arthritis, Whipple’s disease, aspergillus infection, CVID, giant cell arteritis, Crohn’s disease, sarcoidosis or Hodgkin’s lymphoma) [Citation43,Citation63,Citation64]. SAA production from macrophages might contribute to a local inflammatory microenvironment, especially when macrophages are compactly organized in granulomas [Citation58]. Such a granulomatous disease process might somehow facilitate the development of the amyloid nidus as first hit. A second hit might be only increased SAA serum levels by another inflammatory process. An example in which such a “second hit hypothesis” could apply is the case described by Monreal in 1984 in which cholangitis is preceded by tuberculosis earlier in life [Citation65].
Figure 2. Longstanding inflammation and AA amyloid formation. AEF: amyloid enhancing factor; IL1: interleukin-1; IL6: interleukin-6; LPS: lipopolysaccharide; SAA: serum amyloid A protein; TNF: tumour necrosis factor.
Diagnostic approach for patients newly diagnosed with AA amyloidosis
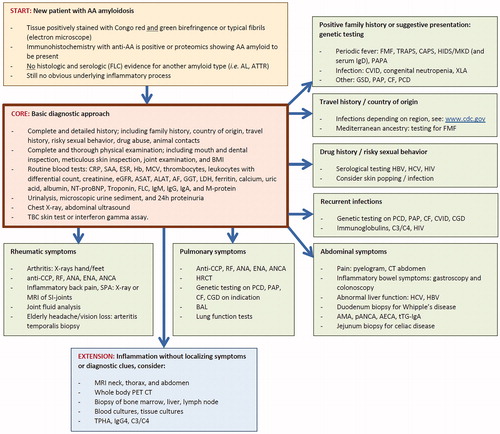
To identify the underlying inflammatory process in newly diagnosed AA amyloidosis a pragmatic diagnostic approach was formulated in , analogous to the ones proposed for idiopathic AA amyloidosis [Citation66] and for fever of unknown origin [Citation67]. A core set should be standard practice as first step in all patients (orange box). In patients without an obvious underlying cause, the second step is individually tailored in seven directions based on specific history or specific symptoms (green boxes). In this respect, it is important to check all the individual boxes, since more than one box can be relevant for a patient. For example, in all patients of Jewish, Turkish, Armenian, Italian, Greek, Spanish or Arabian ancestry familial Mediterranean fever (FMF) should be ruled out as underlying cause, since also asymptomatic cases can develop AA amyloidosis [Citation68]. However, boxes should be screened based on the symptoms present in that particular patient. If these two steps do not reveal the underlying inflammatory process, a more extensive search is justified (blue box). Even after detecting a particular underlying disease the physician in charge must remain critical: if the acute phase response (CRP, SAA) does not disappear after successfully curing the supposed underlying disease (e.g. TBC), a possible other inflammatory process (e.g. bronchiectasis) must be considered.
Figure 3. Flow chart of the diagnostic approach to detect the underlying disease process in a patient with newly diagnosed AA amyloidosis, but still without an obvious cause (yellow box). First step is obtaining a complete history and performing a thorough physical examination, together with a core set of basic laboratory measurements and imaging (orange box). Second step shows the possible investigations in seven directions guided by specific clues obtained from history or from symptoms (green boxes). If localizing symptoms or diagnostic clues remain absent, a full search for the underlying inflammatory process is justified (blue box). AA: amyloid A; AECA: anti-endothelial cell antibody; AF: alkaline phosphatase; AL: amyloid light chain-related; ALAT: alanine amino transferase; AMA: anti-mitochondrial antibody; ANA: antinuclear antibody; ANCA: anti-neutrophil cytoplasmic antibody; anti-CCP: anti-cyclic citrullinated protein; ASAT: aspartate amino transferase; ATTR: amyloid transthyretin-related; BAL: bronchoalveolar lavage; BMI: body mass index; C3/C4: complement factor 3 and 4; CAPS: cryopyrin-associated periodic syndrome; CF: cystic fibrosis; CGD: chronic granulomatous disease; CRP: C-reactive protein; CT: computed tomography; CVID: common variable immunodeficiency disorder; eGFR: estimated glomerular filtration rate; ENA: antibodies against extractable nuclear antigen; ESR: erythrocyte sedimentation rate; FLC: immunoglobulin free light chain; FMF: familial Mediterranean fever; GGT: gamma glutamyl transferase; GSD: glycogen storage disease; Hb: hemoglobin; HBV: hepatitis B virus; HCV: hepatitis C virus; HIDS: hyperimmunoglobulinemia D syndrome; HIV: human immunodeficiency virus; HRCT: high-resolution computed tomography; IgA: immunoglobulin A; IgD: immunoglobulin D; IgG: immunoglobulin G; IgG4: Immunoglobulin G subtype 4; IgM: immunoglobulin M; LDH: lactate dehydrogenase; M-protein: monoclonal protein; MCV: mean corpuscular volume; MKD: mevalonate kinase deficiency; MRI: magnetic resonance imaging; NT-proBNP: N-terminal pro-B-type natriuretic protein; pANCA: perinuclear ANCA; PAP: pulmonary alveolar proteinosis; PAPA: pyogenic sterile arthritis: pyoderma gangrenosum: and acne; PCD: primary ciliary dyskinesia; PET: positron emission tomography; RF: rheumatoid factor; SAA: serum amyloid A protein; TPHA: treponema pallidum haemagglutinase; TRAPS: Tumor necrosis factor receptor-associated periodic syndrome; tTG-IgA: anti-tissue transglutaminase IgA antibodies; XLA: X-linked agammaglobulinemia.
Using the tables in clinical practice
After finding an underlying inflammatory disease in a patient with proven AA amyloidosis, the question rises whether or not the disease is a plausible cause of AA amyloidosis in this patient. lists the strong associations with AA amyloidosis. These 48 diseases are reliable causes if the clinical picture is supportive. This means that the time relations (the duration and course of the underlying disease and the development of amyloidosis disease) are accompanied by longstanding inflammation (elevated SAA or CRP levels) and ideally also by stabilization of the amyloidosis disease after successful treatment (normalizing of SAA and CRP levels). If this is true, the clinical picture supports the particular disease as underlying cause of the AA amyloidosis.
lists 19 diseases with weak associations that may be considered, but only after exclusion of the 48 diseases with a strong association. It should be noticed that more uncertainty is associated with . This uncertainty further increases in that lists 23 diseases lacking a clear association. These diseases are less likely to be causal, but they may be considered if there is overwhelming evidence present and only after exclusion of strong and weak causes. finally lists 60 diseases in which an association seems unlikely. That means that they generally are not real candidates, although a few of them still may be considered (e.g. Schnitzler’s syndrome, ANCA-associated vasculitis or inflammatory pseudotumor) as discussed in the respective paragraph above.
Idiopathic AA amyloidosis
Although gradually more diseases can be tracked down due to improved diagnostic possibilities, an idiopathic rest group remains in which no underlying disease can be detected despite a comprehensive and complete diagnostic search. These cases with an uncertain aetiology are referred to as idiopathic AA amyloidosis [Citation69]. For this review, idiopathic cases were pragmatically defined as: cases in which both amyloid and AA type are proven to be present, other amyloid types made unlikely and in which other associated diseases were ruled out by the authors. The designation idiopathic AA amyloidosis is actually an admission of weakness and the underlying mechanism needs clarification. A first step is to collect these cases and look for characteristics in common [Citation66,Citation70–72]. Possible subgroups may be identified, for example by SAA1 subtyping. Obesity, SAA1.1/1.1 homozygosity and elevated CRP were associated with an increased risk of developing idiopathic AA amyloidosis [Citation73]. However, almost half of the patients with idiopathic amyloidosis in this series [Citation73] (18 of 37) had a body mass index (BMI)>30 and should preferably be classified as obesity-related [Citation46].
Limitations of the study
A first limitation is the absence of many articles published before 1980. The main reason was that a reliable diagnosis of AA amyloid was not possible before the introduction of anti-AA immunohistochemistry in the mid-eighties. Consequently, the frequent development of amyloidosis in some infectious diseases (e.g. syphilis) before the era of antibiotics remains hidden in this review. A second limitation is the exclusion of articles with languages other than English, Dutch, French or German, which may have led to an underestimation of disease associations rare in the USA and Western Europe, but endemic in Mediterranean, African, South American or Asian countries (e.g. malaria and other tropical diseases) or Russia (e.g. Leishmanisis, echinococcus). A third and more fundamental limitation is that a large part of humanity lives in poor and less-privileged circumstances in which amyloidosis and associated diseases simply are not recognized and if recognized will not be published at all. A fourth limitation is the quality of the available case reports. Many cases were reported by authors with little experience in amyloidosis, leading to incomplete case descriptions and data collection. Since amyloidosis is often an unexpected and late problem in the disease course of a patient, there is also not much time left to collect all relevant data for an undisputable case report. A fifth limitation is the requirement of at least two proven cases with AA amyloidosis and a particular disease as condition for a strong association. This may underestimate the presence of AA amyloidosis in very rare diseases. On the other side, coincidence may still be present even when two cases are found, certainly in the more common underlying diseases. The last limitation is the recognition of diseases and disease clusters. A very restricted disease definition will make it difficult to find at least two associated proven cases, whereas a broad disease definition in a cluster will make it easier to find, but the association is in such a case not related to a specific disease. This played a role in the vasculitis and tumour categories. Furthermore, it should be noticed that a strong association of a disease with AA amyloidosis does not necessarily indicate a frequent association in clinical practice (e.g. FMF versus gout). However, this is something that goes beyond the purpose of this manuscript.
A problem that comes along with the above-mentioned limitations is the finding that diseases that are assumed to cause amyloid in general practice (e.g. syphilis or ANCA-associated vasculitis) cannot be listed as a strong or even weak association due to lack of evidence in the literature studied. When viewed from the bright side, this review is just a beginning and adequate new case descriptions can be used to complement or revise the list of causes in a future update. Furthermore, the criteria used can be improved and validated, which is even more desirable for the proposed work-up scheme and criteria of idiopathic AA amyloidosis, because diagnostic possibilities constantly undergo improvement and will be used in a different way in the near future.
Conclusions
AA amyloidosis is caused by different diseases inducing increased SAA serum levels. SAA levels can directly rise by infections, inflammation or malignancies. But SAA levels can also indirectly rise due to increased susceptibility to infections as seen in CVID, cyclic neutropenia and cystic fibrosis, or to an increased risk of chronic inflammation as seen in GSD. Knowing the underlying disease provides an opportunity to look for targeted therapy, since normalizing SAA serum levels by adequately supressing the underlying disease determines the prognosis of AA amyloidosis. Therefore, a pragmatic diagnostic approach was proposed, which can be used in patients with newly diagnosed AA amyloidosis. It provides also diagnostic support for establishing idiopathic AA amyloidosis and for patients in whom an unlikely or unclear associated condition is found and other diseases need to be considered first.
| Abbreviations | ||
| AA amyloidosis | = | inflammation-associated amyloidosis |
| Aβ2M | = | β2-microglobulin-associated amyloid |
| AECA | = | anti-endothelial cell antibody |
| AEF | = | amyloid-enhancing factor |
| AF | = | alkaline phosphatase |
| AIDS | = | acquired immunodeficiency syndrome |
| AL amyloid | = | immunoglobulin light chain-associated amyloid |
| ALAT | = | alanine amino transferase |
| AMA | = | anti-mitochondrial antibody |
| ANA | = | antinuclear antibody |
| ANCA | = | antineutrophil cytoplasmic antibody |
| anti-CCP | = | anti-cyclic citrullinated protein |
| ASAT | = | aspartate amino transferase |
| ATTR | = | transthyretin-associated |
| BAL | = | bronchoalveolar lavage |
| BMI | = | body mass index |
| C3/C4 | = | complement factor 3 and 4 |
| CAPS | = | cryopyrin-associated periodic syndrome |
| CF | = | cystic fibrosis |
| CGD | = | chronic granulomatous disease |
| CRP | = | C-reactive protein |
| CT | = | computed tomography |
| CVID | = | common variable immunodeficiency disorder |
| eGFR | = | estimated glomerular filtration rate |
| ENA | = | antibodies against extractable nuclear antigen |
| ESR | = | erythrocyte sedimentation rate |
| FLC | = | immunoglobulin free light chain |
| FMF | = | familial Mediterranean fever |
| GBCC | = | giant basal cell carcinoma |
| GCA | = | giant cell arteritis |
| GGT | = | gamma glutamyl transferase |
| GIST | = | gastrointestinal stromal tumour |
| GSD | = | glycogen storage disease |
| Hb | = | hemoglobin |
| HBV | = | hepatitis B virus |
| HCV | = | hepatitis C virus |
| HIDS | = | hyperimmunoglobulinemia D syndrome |
| HIV | = | human immunodeficiency virus |
| HRCT | = | high-resolution computed tomography |
| IgA | = | immunoglobulin A |
| IgD | = | immunoglobulin D |
| IgG | = | immunoglobulin G |
| IgG4 | = | immunoglobulin G subtype 4 |
| IgM | = | immunoglobulin M |
| KMnO4 | = | potassium permanganate |
| LDH | = | lactate dehydrogenase |
| M-protein | = | monoclonal protein |
| MCTD | = | mixed connective tissue disease |
| MCV | = | mean corpuscular volume |
| MGUS | = | monoclonal gammopathy with undetermined significance |
| MKD | = | mevalonate kinase deficiency |
| MRI | = | magnetic resonance imaging |
| NT-proBNP | = | N-terminal pro-B-type natriuretic protein |
| pANCA | = | perinuclear ANCA |
| PAP | = | pulmonary alveolar proteinosis |
| PAPA | = | pyogenic sterile arthritis: pyoderma gangrenosum: and acne |
| PCD | = | primary ciliary dyskinesia |
| PET | = | positron emission tomography |
| PMR | = | polymyalgia rheumatica |
| RA | = | rheumatoid arthritis |
| SAA | = | serum amyloid A protein |
| RF | = | rheumatoid factor |
| SAPHO | = | synovitis, acne, pustulosis, hyperostosis and osteitis |
| SDH | = | succinate dehydrogenase |
| SLE | = | systemic lupus erythematosus |
| TBC | = | tuberculosis |
| TNF-α | = | tumour necrosis factor-alpha |
| TPHA | = | treponema pallidum haemagglutinase |
| TRAPS | = | Tumour necrosis factor receptor-associated periodic syndrome |
| tTG-IgA | = | anti-tissue transglutaminase IgA antibodies |
| XLA | = | X-linked agammaglobulinemia |
Causes_of_AA_amyloidosis_Supplementary_file_final_version_31-10-2019.docx
Download MS Word (142.2 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Obici L, Merlini G. AA amyloidosis: basic knowledge, unmet needs and future treatments. Swiss Med Wkly. 2012;142:13580.
- Rokitansky C. Handbuch der speciellen pathologischen Anatomie, volume 3. Wien: Braunmüller und Seidel; 1842. p. 311,324,384.
- Adams W, Dowse TS. Mollities ossium. Tr Path Soc Lond. 1871–1872;23:186–187.
- Pras M, Schubert M, Zucker-Franklin D, et al. The characterization of soluble amyloid prepared in water. J Clin Invest. 1968;47:924–933.
- Terry WD, Page DL, Kimura S, et al. Structural identity of Bence Jones and amyloid fibril proteins in a patient with plasma cell dyscrasia and amyloidosis. J Clin Invest. 1973;52:1276–1281.
- Benditt EP, Eriksen N, Hermodson MA, et al. The major protein of human and monkey amyloid substance: common properties including unusual N-terminal amino acid sequence. FEBS Lett. 1971;19:169–173.
- Levin M, Pras M, Franklin EC. Immunologic studies of the major nonimmunoglobulin protein of amyloid. Identification and partial characterization of a related serum component. J Exp Med. 1973;138:373–380.
- Ein D, Kimura S, Terry WD, et al. Amino acid sequence of an amyloid fibril protein of unknown origin. J Biol Chem. 1972;247:5653–5655.
- Levin M, Franklin EC, Frangione B, et al. The amino acid sequence of a major nonimmunoglobulin component of some amyloid fibrils. J Clin Invest. 1972;51:2773–2776.
- Sletten K, Husby G. The complete amino-acid sequence of non-immunoglobulin amyloid fibril protein AS in rheumatoid arthritis. Eur J Biochem. 1974;41:117–125.
- Sletten K, Husby G, Natvig JB. The complete amino acid sequence of an amyloid fibril protein AA of unusual size (64 residues). Biochem Biophys Res Commun. 1976;69:19–25.
- Møyner K, Sletten K, Husby G, et al. An unusally large (83 amino acid residues) amyloid fibril protein AA from a patient with Waldenström’s macroglobulinaemia and amyloidosis. Scand J Immunol. 1980;11:549–554.
- Skinner M, Pinnette A, Travis WD, et al. Isolation and sequence analysis of amyloid protein AA from a patient with cystic fibrosis. J Lab Clin Med. 1988;112:413–417.
- Liepnieks JJ, Kluve-Beckerman B, Benson MD. Characterization of amyloid A protein in human secondary amyloidosis: the predominant deposition of serum amyloid A1. Biochim Biophys Acta. 1995;1270:81–86.
- Gafni J, Sohar E. Rectal biopsy for the diagnosis of amyloidosis. Am J Med Sci. 1960;240:332–336.
- Cohen AS, Calkins E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature. 1959;183:1202–1203.
- Wright JR, Calkins E, Humphrey RL. Potassium permanganate reaction in amyloidosis. A histologic method to assist in differentiating forms of this disease. Lab Invest. 1977;36:274–281.
- van Rijswijk MH, van Heusden CW. The potassium permanganate method. A reliable method for differentiating amyloid AA from other forms of amyloid in routine laboratory practice. Am J Pathol. 1979;97:43–58.
- Janssen S, Elema JD, van Rijswijk MH, et al. Classification of amyloidosis: immunohistochemistry versus the potassium permanganate method in differentiating AA from AL amyloidosis. Appl Pathol. 1985;3:29–38.
- Said SM, Sethi S, Valeri AM, et al. Renal amyloidosis: origin and clinicopathologic correlations of 474 recent cases. Clin J Am Soc Nephrol. 2013;8:1515–1523.
- Vrana JA, Gamez JD, Madden BJ, et al. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114:4957–4959.
- Dwulet FE, Benson MD. Primary structure of amyloid fibril protein AA in azocasein-induced amyloidosis of CBA/J mice. J Lab Clin Med. 1987;110:322–329.
- Snel FW, Niewold TA, Baltz ML, et al. Experimental amyloidosis in the hamster: correlation between hamster female protein levels and amyloid deposition. Clin Exp Immunol. 1989;76:296–300.
- Li W, Chan SL, Chronopoulos S, et al. Alveolar hydatid cyst (AHC): inflammation-induced reactive gastrointestinal (GI) amyloidosis in AHC-infected mice and chemical characterization of the GI amyloid. Exp Parasitol. 1996;83:1–10.
- Boonpucknavig S, Boonpucknavig V, Tanvanich S, et al. Development of immune-complex glomerulonephritis and amyloidosis in Syrian golden hamsters infected with Opisthorchis viverrini. J Med Assoc Thai. 1992;75:7–19.
- Ovelgönne JH, Landman WJ, Gruys E, et al. Identical amyloid precursor proteins in two breeds of chickens which differ in susceptibility to develop amyloid arthropathy. Amyloid. 2001;8:41–51.
- Hazenberg BP, van Rijswijk MH. Aspects cliniques de l’amylose AA. In: Grateau G, Benson MD, Delpech M, editors. Les amyloses. Paris: Flammarion; 2000. p. 377–427.
- Gillmore JD, Hawkins PN. Pathophysiology and treatment of systemic amyloidosis. Nat Rev Nephrol. 2013;9(10):574–586.
- Papa R, Lachmann HJ. Secondary, AA, Amyloidosis. Rheum Dis Clin N Am. 2018;44(4):585–603.
- Lachmann HJ, Goodman HJ, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007;356(23):2361–2371.
- Benson MD, Buxbaum JN, Eisenberg DS, et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid. 2018;25(4):215–219.
- Linke RP. On typing amyloidosis using immunohistochemistry. Detailled illustrations, review and a note on mass spectrometry. Prog Histochem Cytochem. 2012;47(2):61–132.
- Millucci L, Spreafico A, Tinti L, et al. Alkaptonuria is a novel human secondary amyloidogenic disease. Biochim Biophys Acta. 2012;1822(11):1682–1691.
- Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111–2121.
- Dick J, Kumar N, Horsfield C, et al. AA Amyloidosis in a patient with glycogen storage disorder and progressive chronic kidney disease. Clin Kidney J. 2012;5(6):559–561.
- Wang DQ, Fiske LM, Carreras CT, et al. Natural history of hepatocellular adenoma formation in glycogen storage disease type I. J Pediatr. 2011;159(3):442–446.
- Lee PJ. Glycogen storage disease type I: pathophysiology of liver adenomas. Eur J Pediatr. 2002;161:46–49.
- Kikuchi M, Haginoya K, Miyabayashi S, et al. Secondary amyloidosis in glycogen storage disease type Ib. Eur J Pediatr. 1990;149(5):344–345.
- Shibasaki T, Matsumoto H, Watabe K, et al. A case of renal amyloidosis associated with hepatic adenoma: the pathogenetic role of tumor necrosis factor-α. Nephron. 1997;75(3):350–353.
- Calderaro J, Letouzé E, Bayard Q, et al. Systemic AA amyloidosis caused by inflammatory hepatocellular adenoma. N Engl J Med. 2018;379(12):1178–1180.
- Chhoda A, Jain D, Kumar Daga M, et al. Celiac disease and secondary amyloidosis: a possible causal association? ACG Case Rep J. 2018;5:e24.
- Muzaffar J, Katragadda L, Haider S, et al. Waldenström’s macroglobulinemia associated with serum amyloid A protein amyloidosis: pitfalls in diagnosis and successful treatment with melphalan-based autologous stem cell transplant. Acta Haematol. 2013;130(3):146–149.
- James DG. A clinicopathological classification of granulomatous disorders. Postgrad Med J. 2000;76(898):457–465.
- Mantovani A, Allavena P, Sica A, et al. Cancer-related inflammation. Nature. 2008;454(7203):436–444.
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–889.
- Stankovic Stojanovic K, Georgin-Lavialle S, Poitou C, et al. AA amyloidosis is an emerging cause of nephropathy in obese patients. Eur J Intern Med. 2017;39:18–20.
- Liu M, Li X, Li Y, et al. Rosai-Dorfman disease with features of IgG4-related disease in the breast: cases report and literature review. Asian Pac J Allergy Immunol. 2018;36(1):51–57.
- Lecamwasam A, Roberts V, Hill P. Nephrotic syndrome in a man with Carney-Stratakis syndrome. Pathology. 2015;47(7):705–707.
- Sokabe A, Mizooka M, Sakemi R, et al. Systemic inflammatory syndrome associated with a case of jugular paraganglioma. Intern Med. 2016;55(15):2105–2108.
- Muniz-Pacios L, Morales-Ruiz E, Aguilar F, et al. Chronic renal failure secondary to systemic amyloidosis associated with gastrointestinal stromal tumour. Nefrologia. 2013;33:620–622.
- Overstreet K, Barone RM, Robin HS. Secondary amyloidosis and gastrointestinal stromal tumors. A case report and discussion of pathogenesis. Arch Pathol Lab Med. 2003;127(4):470–473.
- Jaakkola H, Tornroth T, Groop PH, et al. Renal failure and nephrotic syndrome associated with gastrointestinal stromal tumor (GIST) – a rare cause of AA amyloidosis. Nephrol Dial Transplant. 2001;16(7):1517–1518.
- Symmers WS. Amyloidosis complicating actinomycosis. Br Med J. 1973;4(5889):423–424.
- Masterton G. Cardiovascular syphilis with amyloidosis and periods of alternating heart block. Br J Vener Dis. 1965;41:181–185.
- Szymanski FJ, Gecht ML, Fox JM. Chronic lymphatic leukemia with secondary amyloidosis. Arch Dermatol. 1959;80:603.
- Kaynar K, Sukru U, Ozoran Y, et al. Ureterosigmoidostomy and secondary amyloidosis. Health Med. 2011;5:292–294.
- De Buck M, Gouwy M, Wang JM, et al. The cytokine-serum amyloid A-chemokine network. Cytokine Growth Factor Rev. 2016;30:55–69.
- Jumeau C, Awad F, Assrawi E, et al. Expression of SAA1, SAA2 and SAA4 genes in human primary monocytes and monocyte-derived macrophages. PLoS One. 2019;14(5):e0217005.
- Dhillon V, Woo P, Isenberg D. Amyloidosis in the rheumatic diseases. Ann Rheum Dis. 1989;48(8):696–701.
- Obici L, Raimondi S, Lavatelli F, et al. Susceptibility to AA amyloidosis in rheumatic diseases: a critical overview. Arthritis Rheum. 2009;61(10):1435–1440.
- Solomon A, Richey T, Murphy CL, et al. Amyloidogenic potential of foie gras. Proc Natl Acad Sci USA. 2007;104(26):10998–11001.
- Westermark GT, Fändrich M, Westermark P. AA amyloidosis: pathogenesis and targeted therapy. Annu Rev Pathol Mech Dis. 2015;10(1):321–344.
- Rose CD, Neven B, Wouters C. Granulomatous inflammation: the overlap of immune deficiency and inflammation. Best Pract Res Clin Rheumatol. 2014;28(2):191–212.
- Ehrenstein B, Brochhausen C. Differenzialdiagnose granulomatöser Erkrankungen. Z Rheumatol. 2017;76(5):415–424.
- Monreal FA. Pulmonary amyloidosis: ultrastructural study of early alveolar septal deposits. Hum Pathol. 1984;15(4):388–389.
- Lane T, Wechalekar AD, Gillmore JD, et al. Safety and efficacy of empirical interleukin-1 inhibition using anakinra in AA amyloidosis of uncertain aetiology. Amyloid. 2017;24(3):189–193.
- Knockaert DC, Van der Schueren S, Blockmans D. Fever of unknown origin in adults: 40 years on. J Intern Med. 2003;253(3):263–275.
- Peces R, Afonso S, Peces C, et al. Living kidney transplantation between brothers with unrecognized renal amyloidosis as the first manifestation of familial Mediterranean fever: a case report. BMC Med Genet. 2017;18(1):97.
- Pras M, Zaretzky J, Frangione B, et al. AA protein in a case of “primary” or “idiopathic” amyloidosis. Am J Med. 1980;68(2):291–294.
- Blank N, Hegenbart U, Lohse P, et al. Risk factors for AA amyloidosis in Germany. Amyloid. 2015;22(1):1–7.
- Bunker D, Gorevic P. AA amyloidosis: Mount Sinai experience, 1997–2012. Mt Sinai J Med. 2012;79(6):749–756.
- Girnius S, Dember L, Doros G, et al. The changing face of AA amyloidosis: a single center experience. Amyloid. 2011;18(Suppl 1):226–228.
- Blank N, Hegenbart U, Dietrich S, et al. Obesity is a significant susceptibility factor for idiopathic AA amyloidosis. Amyloid. 2018;25(1):37–45.