
ABSTRACT
Melittin, a naturally occurring polypeptide found in bee venom, has been recognized for its potential anti-tumor effects, particularly in the context of lung cancer. Our previous study focused on its impact on human lung adenocarcinoma cells A549, revealing that melittin induces intracellular reactive oxygen species (ROS) burst and oxidative damage, resulting in cell death. Considering the significant role of mitochondria in maintaining intracellular redox levels and ROS, we further examined the involvement of mitochondrial damage in melittin-induced apoptosis in lung cancer cells. Our findings demonstrated that melittin caused changes in mitochondrial membrane potential (MMP), triggered mitochondrial ROS burst (Figure 1), and activated the mitochondria-related apoptosis pathway Bax/Bcl-2 by directly targeting mitochondria in A549 cells (Figure 2). Further, we infected A549 cells using a lentivirus that can express melittin-Myc and confirmed that melittin can directly target binding to mitochondria, causing the biological effects described above (Figure 2). Notably, melittin induced mitochondrial damage while inhibiting autophagy, resulting in abnormal degradation of damaged mitochondria (Figure 5). To summarize, our study unveils that melittin targets mitochondria, causing mitochondrial damage, and inhibits the autophagy-lysosomal degradation pathway. This process triggers mitoROS burst and ultimately activates the mitochondria-associated Bax/Bcl-2 apoptotic signaling pathways in A549 cells.
1. Introduction
Cancer poses a significant global health burden, and this burden is projected to rise in the next two decades [Citation1]. Among various malignancies, lung cancer stands as the foremost cause of cancer-related mortality. There were nearly 2.21 million cases of lung cancer reported, resulting in approximately 1.8 million deaths associated with the disease in 2020 [Citation2]. Lung cancer encompasses distinct subtypes, including non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), with NSCLC constituting 85% of cases [Citation3]. Notably, lung adenocarcinoma (LUAD) represents the most prevalent subtype of NSCLC, exhibiting a 5-year survival rate of merely 15% [Citation4]. Despite significant advancements in lung cancer treatment, such as immunotherapy and targeted therapies, many patients still grapple with the adverse effects of frequent treatment modifications due to primary or acquired drug resistance, leading to tumor recurrence and metastasis [Citation5]. Given the persistent challenges associated with LUAD, the quest for effective antitumor drugs remains an urgent priority in the realm of public health.
With the advancement of antitumor drugs, the research community has discovered that peptides offer several advantages over small molecules and biologics, with a more straightforward design and more remarkable ability to interoperate with the target, as well as desirable immunogenicity and intense tissue penetration. Melittin, a polypeptide derived from bee venom consisting of 26 amino acids, exhibits significant antiviral, antibacterial, anti-inflammatory, and anti-tumor properties [Citation6]. Melittin possesses robust surface activity on lipid membranes, allowing it to interact with membranes and induce membrane fragmentation [Citation6]. In recent years, melittin has shown promising therapeutic effects in various types of tumors, including glioblastoma [Citation7], breast cancer [Citation8] and melanoma [Citation9], and others. Moreover, the therapeutic potential of melittin in lung cancer has been progressively revealed. Our previous study demonstrated that melittin exerts notable anti-tumor activity by inducing a burst of reactive oxygen species (ROS) and oxidative damage in A549 [Citation10]. In human tumor cells, ROS plays a dual role. Elevated ROS can promote tumor signaling and contribute to aggressive tumor growth. However, tumor cells also require efficient removal of accumulated ROS to maintain levels below the toxic threshold. Therefore, strategies that enhance ROS production or inhibit its elimination, leading to the accumulation of intracellular ROS, are considered practical approaches for cancer treatment [Citation11–13].
Mitochondria play a critical role in maintaining cellular oxidative stress balance and serve as the powerhouse for intracellular oxidative respiration, providing the energy necessary for cell growth, proliferation, differentiation, and metabolism [Citation14]. Mitochondria have a double-membrane structure, and the electron transport chain present on the inner mitochondrial membrane (IMM) is the core of intracellular oxidative respiration [Citation15,Citation16]. Mitochondrial damage can lead to impaired function, and ROS burst, activating the mitochondrial apoptosis Bax/Bcl-2 signaling pathway, ultimately leading to apoptosis [Citation17]. Moreover, cancer cells often exhibit heightened mitochondrial metabolism to sustain their high energy demands. Consequently, mitochondria have emerged as an appealing target for therapeutic interventions in oncology. In recent years, there has been raising interest in the application of mitochondrial-targeted therapies in cancer treatment, including photodynamic therapy [Citation18], drug delivery supplementation [Citation19,Citation20], as well as the development of mitochondria-targeted drugs [Citation21,Citation22] and related materials [Citation23].
Autophagy is a crucial metabolic process, preserving normal cellular metabolism by recycling damaged organelles and cellular components. It includes the selective removal of damaged mitochondria, known as mitophagy [Citation24]. Mitophagy is triggered by profound mitochondrial depolarization, primarily resulting from impaired oxidative phosphorylation (OXPHOS) [Citation25]. In the context of tumor development, mitophagy has been found to exhibit double duty. On the one hand, it can contribute to processes such as carcinogenesis, cell migration, maintenance of cancer stemness, and drug resistance. On the other hand, inducing mitophagy with specific pharmacological agents has shown potential for disrupting normal cellular metabolism, eliciting cellular stress responses, and causing genetic damage through augmented mitochondrial dysfunction, thus promoting an antitumor effect [Citation26].
In this study, we examined the impact of melittin on mitochondrial homeostasis and demonstrated that melittin-induced mitochondrial depolarization and impaired mitochondrial autophagy are crucial factors resulting in cancer cell apoptosis.
2. Methods
2.1. Materials
The synthesis of melittin was described in our previous study [Citation10,Citation28]. T25 cell culture flasks were purchased from NEST Biotechnology (Wuxi, China). Cell culture plants were from Jet Biofil (Guangzhou, China). The LC3B and P62 antibodies were performed from CST (Danvers, MA, USA). The Anti-β-Tubulin antibody was acquired from Santa Cruz Biotechnology (CA, USA). Bafilomycin A1 (Baf A1) and 3-methyladenine (3-MA) were obtained from Sangon Biotech (Shanghai, China). MitoSox Red was purchased from Merk (Beijing, China).
2.2. Cell culture and treatment
A549 cells were cultured with 10% Fetal bovine serum (FBS) (CellMax, Beijing, China) and Ham‘s F12 K nutrient medium (Viva Cell, Shanghai, China) as previous description [Citation27]. Cells were dealt with or without 3-MA (or Baf A1) for 2 h, followed by incubation with melittin for 24 h. After treatment, cells were harvested for subsequent experiments.
2.3. RNA sequencing
For transcriptome analysis of the control and melittin-treated group, high throughput sequencing of cDNA using next-generation sequencing (NGS) was performed. Cells were treated with or without melittin (4 μg/ml) for 24 h and collected for sequence testing. RNA integrity and quantity were assessed through the RNA Nano 6000 Assay Kit (Nanogene, Beijing, China) on the Bioanalyzer 2100 system from Agilent Technologies. After mRNA purification, reverse transcription was performed to convert mRNA into cDNA. The cDNA was then subjected to double-stranded DNA synthesis using DNA polymerase and dNTPs. The resulting DNA fragments were treated to convert overhangs into blunt ends using exonuclease and polymerase activities. Adenylation was performed on the 3’ ends of the DNA fragments, followed by ligation of adaptors with hairpin loop structures for hybridization. To obtain cDNA fragments of the desired length (370–420 bp), the library fragments were purified using the AMPure XP system from Beckman Coulter. Subsequently, PCR amplification was carried out, and the PCR product was purified using AMPure XP beads to obtain the final library. After quality control, the libraries were pooled based on their effective concentration and the desired sequencing depth. The pooled libraries were then subjected to sequencing using the Illumina NovaSeq 6000 platform. Four fluorescently labeled dNTPs, DNA polymerase, and adapter primers are introduced into the flow cell of the sequencing platform for amplification. As each complementary strand in every sequencing cluster extends, the incorporation of a labeled dNTP releases a corresponding fluorescent signal. The sequencer captures these fluorescence signals and, with the aid of computer software, translates the light signals into sequencing peaks, thereby obtaining the sequence information of the target fragment.
To ensure the quality and reliability of data analysis, it is necessary to filter the raw data in subsequent steps. All analyses were conducted based on clean data. Download the reference genome and gene model annotation files directly from the genome website. Built an index for the reference genome using HISAT2 (v2.0.5) and aligned the paired-end clean reads to the reference genome using HISAT2 (v2.0.5). FeatureCounts (1.5.0-p3) was used to calculate the read counts mapped to each gene. Subsequently, the FPKM (Fragments Per Kilobase Million) for each gene was computed based on gene length, along with the calculation of read counts mapped to that gene.
As biological replicate samples, the differential expression analysis was performed using the DESeq2 R package (version 1.20.0) on the control and melittin groups, with three biological replicates for each condition. DESeq2 utilizes statistical algorithms based on the negative binomial distribution to identify differential expression in digital gene expression data. The resulting p-values were adjusted for multiple testing using the Benjamini and Hochberg method to control the false discovery rate. Differential gene analysis and differential gene pathway enrichment were performed by NovoMagic v3.0.
2.4. Cell viability assay
A549 cells were planted to 96-well overnight and pretreated with melittin (0, 0.5, 1, 2, 4, 8μg/ml) for 24 h. Cell viability was analyzed by Cell Counting Kit-8 assay (K1018, APExBIO, Houston, USA) and MTT assay. CCK-8 assay was recorded at OD 450 nm. MTT assay method aligned with our previous research [Citation28].
2.5. Measurement of MMP (δψM)
The changes of mitochondrial membrane potential (MMP, ΔψM) induced by melittin were assessed by the JC-1 detection kit. A549 cells were planted into 12-well plates for 1 × 105 cells per. After treatment with or without melittin (0.5, 1, 2, 4, 8μg/ml) for 24 h, cells were incubated with JC-1 for 20 min. Subsequently, we employed fluorescence microscopy (IX71, Olympus) to capture images.
2.6. Measurement of mitoROS
A549s were pretreated with or without Baf A1 for 2 hr and then incubated with melittin (0, 0.5, 1, 2, 4, 8μg/ml) for 24 h. Then, MitoSox Red labeled mitoROS was observed by fluorescent microscope (IX71, Olympus).
2.7. Western blot assay
A549 cells were pretreated with or without 3-MA or Baf A1 for 2 h. Following the pretreatment, cells were incubated with melittin for 24 h. After the incubation, cells were collected and lysed on ice using IP lysate containing 5% PMSF. The protein concentration was determined using the BCA assay kit. SDS-PAGE (10% gel) resolved an equal amount of protein and transferred onto nitrocellulose membranes through a transfer system. The membrane was blocked with 5% skimmed milk powder. Subsequently, it was incubated overnight with primary antibodies at 4°C, followed by a 1-hour incubation with secondary antibodies at room temperature. The detailed steps followed the methods described in our previous study [Citation14].
2.8. Transmission electron microscope (TEM)
Cells were pretreated with or without melittin (4μg/ml) for 24 h. Following the pretreatment, cells were incubated with melittin for 24 h. The observation was conducted using transmission electron microscopy to examine intracellular mitochondrial damage and melittin-induced morphological alterations in A549 cells. Initially, fixation was achieved with 3% glutaraldehyde, followed by sample fixation in the 1% osmium tetroxide solution. Subsequently, samples were subjected to stepwise ethanol dehydration and final dehydration in acetone. After infiltration and embedding, ultrathin nanoscale sections were prepared. These sections were stained with uranyl acetate for 10 min, followed by 2-minute staining with lead citrate at room temperature. Talos F200C was used to capture images (Thermo Scientific, USA).
2.9. Measurement of ATP
Cells were planted into 6-well plates overnight and dealt with or without melittin (4 μg/ml) for 24 h. Aspirate the culture medium and lyse the cells by adding lysis solution at a ratio of 200 µl of lysis solution per well of a 6-well plate. The cells were centrifuged at 12,000 g for 5 min, leaving the sediment for subsequent assays. The test solution is configured according to the manufacturer's agreement (Beyotime, S0026) and performed with the luminometer.
2.10. Measurement of apoptosis
A549 cells were pretreated with or without 3-MA or Baf A1, then incubated with melittin (0, 4μg/ml) for 24 h. Apoptosis was assessed by the Annexin-V and PI apoptosis assay kit. According to the manufacturer's protocol, we added the probes into the cells and captured images under the fluorescence microscope (IX71, Olympus).
2.11. Analysis of autophagy (mitophagy) levels
A549 were cultured in a 6-well plate overnight, followed by transfection with GFP-LC3 adenovirus (40 MOI) for 24 h. Subsequently, cells were dealt with or without Baf A1 for 2 h and melittin (0, 2, 4 μg/ml) for another 24 h. Afterward, intracellular mitochondria were labeled with Mitotracker Red. The fluorescent signals of intracellular mitochondria were detected by confocal microscopy (Nikon, A1R + Ti2-E).
2.12. Statistical analysis
All experimental data were collected from a minimum of three independent experiments. Statistical analysis was conducted by calculating the mean ± standard deviation (SD). Prism 9.0 Software (GraphPad Software), Image j 1.53 and Origin 8.0 Software were used for data representation and analysis. The student's t-test was employed for statistical analysis. Statistical significance was established at a threshold of p < 0.05.
3. Results
3.1. Melittin induces mitochondrial dysfunction and exerts antitumor effects
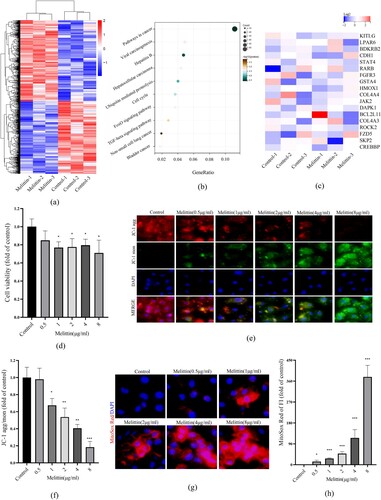
To explore the effect of melittin on A549 cells at the transcriptional level, RNA sequencing analysis was performed to examine the mRNA levels after melittin treatment. According to the KEGG analysis, we found some cancer-related pathways have significant downregulation upon melittin treatment, such as pathways in cancer, cell cycle, FoxO signaling pathway and TGF-beta signaling pathway ((a-b)). In the KEGG analysis of pathways in cancer, cancer-related genes such as kitlg, lpar6, bdkrb2, cdh1, stat4, rarb, fgfr3, gsta4, hmox1, col4a4, jak2, dapk1, bcl2l11, col4a3, rock2, fzd5, skp2, crebbp and others were significantly decreased ((c)). Moreover, the results of CCK8 detection showed that melittin has a significant tumor-killing effect in a dose-dependent way ((d)). These findings were in line with our previous investigations and further support the potential therapeutic function of melittin in cancer. Our previous studies have demonstrated that melittin-induced intracellular ROS burst significantly affects the anti-lung cancer efficiency. Considering that mitochondria are the primary suborganelle of ROS generation and oxidative respiration, we speculate that melittin may have an impact on mitochondrial function. To explore the function of mitochondria in melittin-treated cells, we assessed mitochondrial membrane potential (MMP) by conducting the JC-1 assay to investigate this. When the level of mitochondrial membrane potential is high, JC-1 molecules aggregate within the mitochondrial matrix, forming polymerized structures (J-aggregates) that emit red fluorescence. When the MMP level is low, JC-1 does not aggregate within the mitochondrial matrix and exists as monomers, emitting green fluorescence. Consequently, changes in mitochondrial membrane potential can be detected through the shift in fluorescence colors. The relative ratio of red to green fluorescence is commonly used to quantify the degree of mitochondrial depolarization. In (e-f), we have quantified the changes in JC-1 aggregation/monomer ratio and observed that, with an increasing concentration of melittin, the MMP level decreased. Furthermore, we utilized the MitoSox Red probe to label mitoROS and observed a substantial increase in mitochondrial ROS production upon melittin treatment ((g-h)). Collectively, these findings indicate that melittin may exert anti-cancer effects by affecting mitochondrial function.
Figure 1. Melittin induces mitochondrial dysfunction and exerts antitumor effects. (a) Cells were treated with or without melittin (4 μg/ml) for 24 h. RNA-sequence analysis. Heatmaps of differential genes in Melittin and Control groups. (b) Pathway enrichment analysis of down-regulated genes. (c) Heatmap of genes of pathways in cancer. (d) Cell proliferation analysis was tested by the CCK8 assay kit. (f) JC-1 assay kit was used to detect the mitochondrial membrane potential in A549. (h) Statistics of JC-1. (g) MitoSox Red was used to analyze mitoROS levels of A549 cells. (h) Analysis of MitoSox Red. *p < 0.05, **p < 0. 01, ***p < 0.001 versus the Control.
3.2. Melittin targets binding to mitochondria and activates Bax/Bcl-2 apoptotic signal pathways
Mitochondrial dysfunction can activate mitochondrial apoptosis-related proteins. The interaction between mitochondrial membrane protein Bcl-2 and Bax can induce the typical mitochondrial apoptosis-inducing systems. We found that the high concentration of melittin triggered the Bax/Bcl-2 apoptotic system activation ((a-b)), suggesting that melittin does induce mitochondrial damage and may trigger apoptosis. Moreover, under high magnification electron microscopy, we visually observed irregular swelling of mitochondria, as well as the fracturing and fuzziness of their cristae ((c)). Additionally, the mitochondria's ability to produce ATP was also inhibited, as shown in (d). Consistent with these experimental results, RNA-sequencing analysis indicated that the transcription of genes related to mitochondrial function, such as minos1, timm10b, mt-tt, mtg1, mt-nd2, mt-co1, mt-cyb, mt-nd1, mt-co3, mrps35 and so on, were significantly reduced after melittin dealt with ((e)). These genes are closely related to the oxidative respiration function of mitochondria. Considering melittin's excellent membrane philic properties, we speculated that melittin-induced mitochondrial dysfunction might be attributed to binding to mitochondria. To confirm our suspicions, we infected A549 cells with Myc-tagged melittin lentivirus and subsequently labeled the cellular mitochondria with Mito-Tracker, and it turned out that melittin can directly target mitochondria in A549 ((f)). These results indicate that melittin may induce mitochondrial dysfunction by directly binding to the mitochondria.
Figure 2. Melittin targets binding to mitochondria and disrupts autophagy flux in A549 cells. (a) Western blot was used to test the level of Bcl-2 and Bax. (b) Statistics of Bax/Bcl-2. (c) Mitochondria morphology was observed with the electron microscope. (d) Statistics of ATP levels. (e) Heat map of mitochondrial function-related downregulated genes in RNA-seq analysis. (f) Cells were transfected with Melittin-Myc lentivirus for 24 h and then labeled with Mito Tracker Green, observed under the confocal microscope. (g) A549 cells were treated with or without melittin (0.5, 1, 2, 4, 8μg/ml) for 24 h. Western blotting was used to autophagy detect the marker protein LC3 and P62. (h) Statistics of P62 levels. (i) Statistics of LC3 alternation. *p < 0.05, **p < 0. 01, ***p < 0.001 versus the Control.
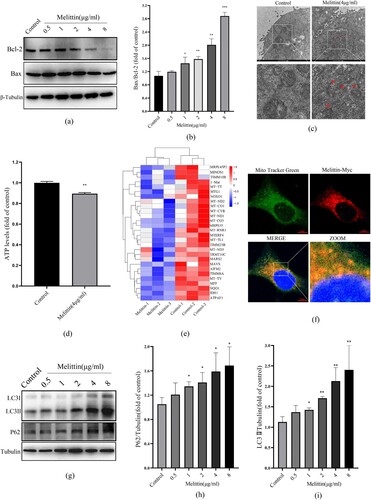
3.3. Melittin induces apoptosis but inhibits autophagy flux in A549 cells
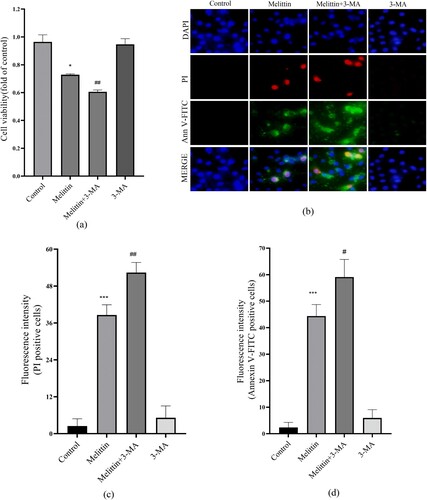
Autophagy is a process charged with removing and recycling dysfunctional organelles and cellular components, including damaged mitochondria. In light of this, we conducted additional investigations to examine the influence of melittin on autophagy. Our findings revealed that melittin treatment significantly increased the conversion of LC3I to LC3II and the accumulation of P62, indicating a substantial inhibition of autophagy flux in A549 cells ((g-i)). To elucidate the connection between altered autophagy levels and melittin-induced cell death, we employed 3-MA to inhibit autophagy. Based on the MTT assay and Annexin V-FITC and PI staining results, it was found that autophagy inhibition 3-MA exacerbated apoptosis and cell death in melittin-treated A549 cells (). In summary, our findings powerfully demonstrate that autophagy serves a significant function in melittin-induced apoptosis.
Figure 3. 3-MA aggravated the cell viability and apoptosis induced by melittin. (a) Cells were pretreated with 3-MA for 2 h and then treated with melittin (4 μg/ml) for 24 h. MTT assay was used to detect cell viability. (b) Detection of apoptosis. (c) Statistics of PI-positive cells. (d) Statistics of Annexin V-FITC-positive cells. *p < 0.05, **p < 0. 01, ***p < 0.001 versus the Control. #p < 0.05, ##p < 0. 01 versus the Melittin group.
3.4. Melittin leads to apoptosis and oxidative stress by inhibiting mitophagy
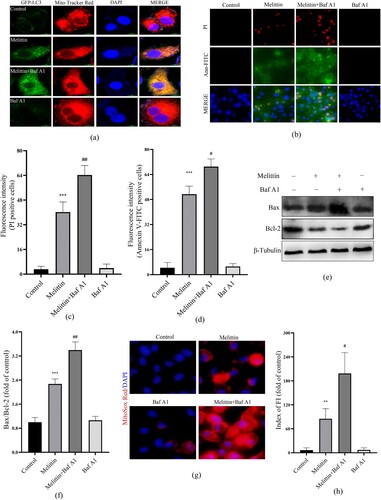
Damaged mitochondria can be targeted by the autophagy system, transferring to lysosomes for degradation, this process helps maintain mitochondrial quality control, which is called mitophagy. Mitochondrial membrane potential (MMP) depolarization is a potential trigger of mitophagy. Consequently, we examined the interplay between mitochondria and autophagy in the presence of melittin. Using GFP-LC3 adenovirus transfected cells and Mito Tracker Red labeled, we observed a marked augmentation in the formation of mitochondrial-autophagosome fusions (yellow puncta) in the melittin group, with a dose-dependent effect ((a)). As a pretreatment, we further employed a late autophagy inhibitor Baf A1 to explore the interaction between mitochondria and autophagy flux. It turned out that Baf A1 resulted in a significant accumulation of fusion of mitochondria and autophagosomes within A549 cells ((a)). Furthermore, the accumulation of damaged mitochondria triggered a subsequent elevation in mitoROS levels ((g)), activation of the Bax/Bcl-2 apoptotic system ((e-f)), leading to apoptosis and cell death ((b-d)).
Figure 4. Melittin resulted in increased intracellular mitochondria-autophagosome fusion. (a) Cells were pre-transfected with the GFP-LC3 virus for 24 h and then incubated with melittin (2,4 μg/ml) for another 24 h. After that cells were labeled with Mito tracker Red for 15 min and analyzed at the confocal fluorescence microscope. Yellow dots show mitophagy in A549 cells. (b) Schematic diagram depicting the induction of mitophagy dysfunction and apoptosis by melittin.
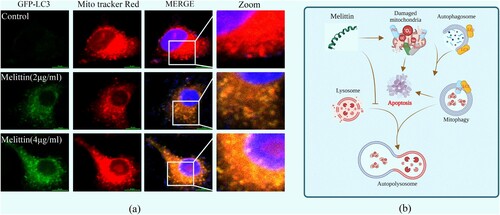
Figure 5. Baf A1 aggravates mitochondria-autophagosome fusion and cytotoxicity induced by melittin. (a) Cells were transfected to the GFP-LC3 virus for 24 h and pretreated with or without Baf A1 for 2 h before being treated with melittin. Final by Mito tracker Red and photographed under the confocal fluorescence microscope. (b) Apoptosis was analyzed by Annexin V-FITC and PI staining. (c) and (d) Statistics of PI and Annexin V-FITC positive staining. (e) cells were pretreated with or without Baf A1 for 2 h and induced by melittin for another 24 h. The protein levels of Bax and Bcl-2 were detected by Western blot assay. (f) Analysis of Bax/Bcl-2. (g) MitoROS levels of A549 cells. (h) Fluorescence intensity of MitoSox Red. *p < 0.05, **p < 0. 01, ***p < 0.001 versus the Control. #p < 0.05, ##p < 0. 01 versus the Melittin group.
4. Discussion
Previous studies have discovered that melittin, a peptide derived from Apis mellifera, possesses an amphiphilic sequence that allows it to interact with lipid bilayers [Citation29]. As research has progressed, it has become evident that melittin not only regulates cell membrane permeability through synergistic effects but also induces functional changes in cells, such as inhibiting ion channels through calmodulin-related pathways [Citation30]. Especially in recent years, melittin, a natural small-molecule polypeptide, has gained attention as a potential anti-tumor drug due to its favorable modification properties and low synthesis cost. Our previous studies have proven that melittin is capable of inducing apoptosis in A549 cells, which is closely related to intracellular reactive oxygen species (ROS) burst and oxidative stress damage [Citation10], and this process was also confirmed in the study by Yu et al,. [Citation31]. However, the precise mechanism underlying melittin-induced intracellular ROS burst and apoptosis remains elusive. Mitochondria, often referred to as the cell's powerhouses, play a crucial role in oxidative respiration, with the electron transport chain on the inner mitochondrial membrane (IMM) serving as the primary source of endogenous ROS production. It is notable that we found that melittin did induce mitochondrial membrane potential (MMP) depolarization and ROS burst in mitochondria, which confirms our suspicion to some extent ().
Decreased MMP causes numerous changes in mitochondrial function, including mitoROS burst, mitophagy, and activation of mitochondrial apoptosis-related signals. Previous studies by Kong et al. found that melittin activates caspase 3 and induces apoptosis in SGC-7901 cells [Citation32]. Within the Bcl-2 family, Bcl-2 and Bax are the most critical regulators of apoptosis known to date and are essential regulatory systems upstream of caspase 3. Upon receiving apoptotic signals, Bax translocates to the mitochondrial membrane, where it forms homodimers or multimers, triggering the formation of permeable pores within the mitochondrial membrane, referred to as mitochondrial permeability transition (PT) pores, disrupting intra-membrane ion and protein concentration differences, and further destroying mitochondrial function. In contrast, Bcl-2 can prevent Bax-mediated damage to the mitochondrial membrane by forming heterodimers with Bax. Our findings demonstrate that melittin activates the Bcl-2/Bax apoptotic pathway associated with mitochondria in a dose-dependent manner ((a-b)). This suggests that melittin activates the mitochondrial apoptotic system Bax/Bcl-2, thus implicating it in the induction of apoptosis. Furthermore, morphological damage of mitochondria, a decrease in mitochondria ATP production capacity, and the downregulation of mitochondrial functional genes provide additional evidence supporting melittin-induced mitochondrial damage ((c-e)). Notably, given the excellent membrane-philic properties of melittin, we hypothesized that melittin-induced mitochondrial dysfunction might involve the direct disruption of mitochondrial membranes. To substantiate our hypothesis, we confirmed that melittin can bind directly to mitochondrial membranes by testing its binding through the expression of melittin fused with Myc tags.
Mitophagy is a crucial component of mitochondrial quality control, which maintains intracellular energy metabolic homeostasis by degrading damaged mitochondria. Mitochondrial membrane depolarization and damage can initiate mitochondrial autophagy, which involves many proteins, including PINK, parkin, P62, BNIP3, LC3 and others. When mitochondria are dysfunctional, MMP is decreased, causing the inability of PINK1 to enter the IMM and its accumulation on the outer mitochondrial membrane (OMM). Accumulated PINK1 activates and recruits the key protein of mitophagy, Parkin, which subsequently mediates the ubiquitination of mitochondrial substrates, including P62, which can further recruit and bind to LC3 on the outer membrane of autophagosomes. Upon the fusion of autophagosomes and lysosomes, damaged mitochondria as well as P62 proteins are degraded. In this study, we found that melittin significantly induced LC3I transforming to LC3II, resulting in a bunch of P62 (). The significantly elevated fusion level of mitochondria with autophagosomes provides evidence that autophagy flux was blocked, which also implies that the degradation of damaged mitochondria was blocked ((a)). Furthermore, dealt with the autophagy inhibitor 3-MA enhanced the inhibitory effect triggered by melittin on cell viability and promoted apoptosis (), providing further evidence that disruption of autophagy serves a pivotal function in the antitumor effect of melittin. To specifically elucidate the function of the mitochondrial autophagy pathway in apoptosis, we pretreated cells with Baf A1, which inhibits the fusion of autophagosomes and lysosomes. As a result, the blockage of autophagy flux by Baf A1 led to the further accumulation of damaged mitochondria within the cells (), resulting in an increase in mitoROS levels and the subsequent activation of the Bax/Bcl-2 apoptotic system, ultimately triggering apoptosis ((b-h)).
Overall, our study indicates that melittin is a mitochondrial membrane-bound peptide that leads to functional and morphological disorders such as impaired mitochondrial ATP production, mitochondrial membrane depolarization, and mitoROS burst to some extent. Additionally, this binding action may cause impaired mitochondrial autophagy, and the amassing of dysfunctional mitochondria further participates in activating the intracellular apoptosis-related pathway Bax/Bcl-2 system, which ultimately exacerbates apoptosis. This suggests that melittin is a promising antitumor drug for application, but we should also note that its immunogenicity due to its amphiphilic nature demands further exploration and research.
Acknowledgments
The authors are grateful to the Core Facility of the School of Life Sciences, Lanzhou University (Gansu province, China).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
Additional information
Funding
References
- Kocarnik JM, Compton K, Dean FE, et al. Cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life years for 29 cancer groups from 2010 to 2019: a systematic analysis for the global burden of disease study 2019. JAMA Oncol. 2022;8:420–444. doi:10.1001/jamaoncol.2021.6987
- Sung H, Ferlay J, Laversanne M, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi:10.3322/caac.21660
- Howlader N, Forjaz G, Mooradian MJ, et al. The effect of advances in lung-cancer treatment on population mortality. N Engl J Med. 2020;383:640–649. doi:10.1056/NEJMoa1916623
- Seguin L, Durandy M, Feral CC. Lung adenocarcinoma tumor origin: a guide for personalized medicine. Cancers (Basel). 2022;14. doi:10.3390/cancers14071759
- Tian Y, Xu L, Li X, et al. SMARCA4: current status and future perspectives in non-small-cell lung cancer. Cancer Lett. 2023;554(Feb 1):216022. doi:10.1016/j.canlet.2022.216022
- Wang A, Zheng Y, Zhu W, et al. Melittin-based nano-delivery systems for cancer therapy. Biomolecules. 2022;12(1):118. doi:10.3390/biom12010118
- Ertilav K, Nazıroğlu M. Honey bee venom melittin increases the oxidant activity of cisplatin and kills human glioblastoma cells by stimulating the TRPM2 channel. Toxicon. 2023;222:106993. doi:10.1016/j.toxicon.2022.106993
- Daniluk K, Lange A, Wójcik B, et al. Effect of melittin complexes with graphene and graphene oxide on triple-negative breast cancer tumors grown on chicken embryo chorioallantoic membrane. Int J Mol Sci. 2023;24. doi:10.3390/ijms24098388
- Jeong C, Kim J, Han IH, et al. Melittin derived peptide-drug conjugate, M-DM1, inhibits tumor progression and induces effector cell infiltration in melanoma by targeting M2 tumor-associated macrophages. Front Immunol. 2023;14:1178776. doi:10.3389/fimmu.2023.1178776
- Li X, Zhu S, Li Z, et al. Melittin induces ferroptosis and ER stress-CHOP-mediated apoptosis in A549 cells. Free Radic Res. 2022;56:398–410. doi:10.1080/10715762.2022.2131551
- Tang JLY, Moonshi SS, Ta HT. Nanoceria: an innovative strategy for cancer treatment. Cell Mol Life Sci. 2023;80:46. doi:10.1007/s00018-023-04694-y
- Perillo B, Di Donato M, Pezone A, et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med. 2020;52:192–203. doi:10.1038/s12276-020-0384-2
- Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. 2018;80:50–64. doi:10.1016/j.semcdb.2017.05.023
- Zhu S, Li X, Wu F, et al. Blue light induces skin apoptosis and degeneration through activation of the endoplasmic reticulum stress-autophagy apoptosis axis: protective role of hydrogen sulfide. J Photochem Photobiol B. 2022;229:112426. doi:10.1016/j.jphotobiol.2022.112426
- Murray AJ, Horscroft JA. Mitochondrial function at extreme high altitude. J Physiol. 2016;594:1137–1149. doi:10.1113/JP270079
- Pernas L, Scorrano L. Mito-Morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol. 2016;78:505–531. doi:10.1146/annurev-physiol-021115-105011
- Wang Y, Chi H, Xu F, et al. Cadmium chloride-induced apoptosis of HK-2 cells via interfering with mitochondrial respiratory chain. Ecotoxicol Environ Saf. 2022;236:113494. doi:10.1016/j.ecoenv.2022.113494
- Yaqoob MD, Xu L, Li C, et al. Targeting mitochondria for cancer photodynamic therapy. Photodiagnosis Photodyn Ther. 2022;38:102830. doi:10.1016/j.pdpdt.2022.102830
- Geng Y, Xiang J, Shao S, et al. Mitochondria-targeted polymer-celastrol conjugate with enhanced anticancer efficacy. J Control Release. 2022;342:122–133. doi:10.1016/j.jconrel.2022.01.002
- Zhang YM, Xia M, Ao R, et al. Smart design of mitochondria-targeted and ROS-responsive CPI-613 delivery nanoplatform for bioenergetic pancreatic cancer therapy. Nanomaterials (Basel). 2021;11.
- Capeloa T, Krzystyniak, d'Hose, et al. Mitoq inhibits human breast cancer cell migration, invasion and clonogenicity. Cancers (Basel). 2022;Mar 16;14(6):1516. doi:10.3390/cancers14061516
- Kalyanaraman B. Exploiting the tumor immune microenvironment and immunometabolism using mitochondria-targeted drugs: challenges and opportunities in racial disparity and cancer outcome research. Faseb j. 2022;36:e22226. doi:10.1096/fj.202101862R
- Luo Z, Gao Y, Duan Z, et al. Mitochondria-targeted self-assembly of peptide-based nanomaterials. Front Bioeng Biotechnol. 2021;9:782234. doi:10.3389/fbioe.2021.782234
- Denisenko TV, Gogvadze V, Zhivotovsky B. Mitophagy in carcinogenesis and cancer treatment. Discov Oncol. 2021;12:58. doi:10.1007/s12672-021-00454-1
- Szczepanowska K, Trifunovic A. Tune instead of destroy: how proteolysis keeps OXPHOS in shape. Biochim Biophys Acta Bioenerg. 2021;1862:148365. doi:10.1016/j.bbabio.2020.148365
- Qiu YH, Zhang TS, Wang XW, et al. Mitochondria autophagy: a potential target for cancer therapy. J Drug Target. 2021;29:576–591. doi:10.1080/1061186X.2020.1867992
- Song J, Huang S, Zhang Z, et al. SPA: a peptide antagonist that acts as a cell-penetrating peptide for drug delivery. Drug Deliv. 2020;27:91–99. doi:10.1080/10717544.2019.1706669
- Zhu S, Li X, Dang B, et al. Lycium Barbarum polysaccharide protects HaCaT cells from PM2.5-induced apoptosis via inhibiting oxidative stress, ER stress and autophagy. Redox Rep. 2022;27:32–44. doi:10.1080/13510002.2022.2036507
- Kleinschmidt JH, Mahaney JE, Thomas DD, et al. Interaction of bee venom melittin with zwitterionic and negatively charged phospholipid bilayers: a spin-label electron spin resonance study. Biophys J. 1997;72:767–778. doi:10.1016/S0006-3495(97)78711-3
- Ridgway Z, Picciano AL, Gosavi PM, et al. Functional characterization of a melittin analog containing a non-natural tryptophan analog. Biopolymers. 2015;104:384–394. doi:10.1002/bip.22624
- Yu R, Wang M, Wang M, et al. Melittin suppresses growth and induces apoptosis of non-small-cell lung cancer cells via down-regulation of TGF-β-mediated ERK signal pathway. Braz J Med Biol Res. 2020;54:e9017. doi:10.1590/1414-431x20209017
- Kong GM, Tao WH, Diao YL, et al. Melittin induces human gastric cancer cell apoptosis via activation of mitochondrial pathway. World J Gastroenterol. 2016;22:3186–3195. doi:10.3748/wjg.v22.i11.3186