
ABSTRACT
Objectives
Intestinal ischemia-reperfusion (I/R) injury is a multifactorial and complex clinical pathophysiological process. Current research indicates that the pathogenesis of intestinal I/R injury involves various mechanisms, including ferroptosis. Methane saline (MS) has been demonstrated to primarily exert anti-inflammatory and antioxidant effects in I/R injury. In this study, we mainly investigated the effect of MS on ferroptosis in intestinal I/R injury and determined its potential mechanism.
Methods
In vivo and in vitro intestinal I/R injury models were established to validate the relationship between ferroptosis and intestinal I/R injury. MS treatment was applied to assess its impact on intestinal epithelial cell damage, intestinal barrier disruption, and ferroptosis.
Results
MS treatment led to a reduction in I/R-induced intestinal epithelial cell damage and intestinal barrier disruption. Moreover, similar to treatment with ferroptosis inhibitors, MS treatment reduced ferroptosis in I/R, as indicated by a decrease in the levels of intracellular pro-ferroptosis factors, an increase in the levels of anti-ferroptosis factors, and alleviation of mitochondrial damage. Additionally, the expression of Nrf2/HO-1 was significantly increased after MS treatment. However, the intestinal protective and ferroptosis inhibitory effects of MS were diminished after the use of M385 to inhibit Nrf2 in mice or si-Nrf2 in Caco-2 cells.
Discussion
We proved that intestinal I/R injury was mitigated by MS and that the underlying mechanism involved modulating the Nrf2/HO-1 signaling pathway to decrease ferroptosis. MS could be a promising treatment for intestinal I/R injury.
Introduction
Ischemia-reperfusion (I/R) injury refers to secondary damage to tissues or organs caused by the restoration of blood supply after a period of ischemia [Citation1], and it is a common pathophysiological damage in gastrointestinal surgery, trauma, intestinal torsion, vascular thrombosis, or incarcerated hernia [Citation2]. I/R injury leads to the excessive production of free radicals, which in turn triggers severe inflammatory responses and induces the overproduction of reactive oxygen species (ROS), thereby exacerbating tissue damage [Citation3]. Intestinal I/R injury often leads to intestinal bacterial translocation, endotoxemia and a massive release of inflammatory cytokines; this not only exacerbates damage to the intestine but also leads to damage to other organs, such as the liver, kidneys, and lungs, causing systemic multiple organ dysfunction syndrome and even death [Citation4]. There are few treatment options available because of the intricate nature of intestinal I/R injury.
Ferroptosis is a different form of cell death triggered by oxidative stress and iron-mediated lipid peroxidation. The major features include cell membrane damage, bubbling, mitochondrial crista shrinking, and increased density of the mitochondrial double membrane [Citation5], which is related to an imbalance in the iron-mediated cellular redox system, leading to metabolic disorders of toxic phospholipid hydroperoxides (PLOOH) and thereby causing damage to the cell membrane [Citation6]. Yang Li et al. [Citation7] confirmed that inhibiting ACSL4 could improve ferroptosis caused by I/R, reducing intestinal damage. Feng et al. [Citation8] reported that inhibiting the generation of ROS and the accumulation of Fe2 + during intestinal IR injury can maintain mitochondrial function and reduce the occurrence of ferroptosis, improving intestinal barrier function. Therefore, inhibiting ferroptosis during intestinal I/R injury may be a potential strategy for improving intestinal barrier function.
Methane is a ubiquitous and essentially nontoxic gas. In humans, carbohydrates in the gastrointestinal tract can produce a large amount of methane through the metabolism of methanogenic microorganisms [Citation9]. Recent studies have shown that methane can be formed through nonmicrobial pathways in all organisms and is closely related to ROS-induced oxidative stress [Citation10]. Methane plays an important role through its anti-inflammatory, antioxidant, and antiapoptotic effects. Methane can significantly reduce the area of myocardial infarction, lower myocardial enzyme levels, and alleviate inflammation and oxidative stress following myocardial infarction [Citation11]. In renal I/R injury, MS treatment improves levels of blood creatinine and blood urea nitrogen (BUN) and reduces pathological conditions in renal tissues by decreasing oxidative stress, cell apoptosis, and inflammation [Citation12]. Similarly, MS plays a crucial role in alleviating hepatic I/R injury by reducing cell apoptosis, inflammatory responses, and enhancing the activity of antioxidant enzymes [Citation13]. Boros et al. [Citation14] reported that adding 2.5% methane to air with a normal oxygen content significantly reduced intestinal I/R injury, which was associated with reduced levels of leukocyte activation and reduced formation of oxygen – and nitrogen-centered free radicals. A study also revealed that inhaling exogenous methane (2.2%) with a normal oxygen content maintained the structure of the intestinal mucosal surface, reduced epithelial permeability, improved local microcirculation, and reduced the generation of reactive oxygen species (ROS) in a rat model of intestinal I/R [Citation15]. Although methane inhalation is efficient and convenient, the use of methane as a flammable and explosive gas poses safety hazards. Therefore, methane can be dissolved in saline water and subjected to high pressure (0.4 MPa, 8 hours) to produce methane saline for subsequent intervention. Our previous studies verified that MS treatment can ameliorate sepsis-induced lung, liver and intestinal damage through its anti-inflammatory and antioxidant effects [Citation16, Citation17]. However, there are few studies on the relationship between MS and intestinal I/R injury. Therefore, in the present study we explored the effect of MS on ferroptosis-mediated intestinal IR injury, providing a new therapeutic method for intestinal I/R injury.
Materials and methods
Experimental animals
Male C57BL/6 mice aged 6–8 weeks and weighing 18–22 g were purchased from Xi'an Jiaotong University. All mice were raised in cages with free access to water and food at a temperature of 25 °C under a 12-hour light/12-hour dark cycle and a relative humidity of approximately 55% and were acclimated and quarantined before the experiments. The animal protocols were designed to minimize discomfort and pain in the mice. All animal experiments followed the standards of the China Animal Protection and Use Committee. The animal procedures used in this study were approved, reviewed, and monitored by the Institutional Animal Care and Use Committee of Xi'an Jiaotong University.
Methane saline preparation
Methane saline (MS) and methane culture medium (MM) were prepared as described in previous studies [Citation18]. Methane gas was pressurized (0.4 MPa-0.5 MPa) for 4 hours to dissolve it in 0.9% (w/v) saline and cell culture medium, producing supersaturated MS and MM. The concentrations ranged from approximately 1.2-1.5 mmol/l and 1.4-1.8 mmol/l, respectively. The concentrations were estimated by gas chromatography.
Experimental design
The intestinal I/R mouse model was generated as previously described [Citation19]. After anesthesia with 1% (w/v) pentobarbital sodium, laparotomy was conducted to locate the superior mesenteric artery (SMA), which was then clamped with a nontraumatic vascular clamp for 30, 60, or 90 minutes before releasing the clamp to restore the blood supply for 120 minutes. The sham group received the same surgical treatment but without clamping the SMA. The Caco-2 cell line was purchased from Shanghai Institute of Biochemistry and Cell Biology. Oxygen-glucose deprivation/reperfusion (OGD/R) was used to establish an I/R model in vitro. The cells were cultured under 95% N2 and 5% CO2 in glucose-free medium for 2, 4, or 6 hours to establish anoxia, followed by reoxygenation in glucose-containing medium under 5% O2 and 5% CO2 for 2 hours.
To observe the effects of MS on ferroptosis during I/R injury, the mice were divided into four groups: the sham group, I/R group, I/R + MS group and I/R + Fer-1 group (n = 10, for each group). Ferrostatin-1 was used as a positive control. Fer-1 (5 mg/kg) and MS (10 ml/kg) were administered intraperitoneally to different groups of mice after 60 minutes of ischemia and before reperfusion, followed by euthanasia after 120 minutes of reperfusion and collection of intestinal tissues and blood samples. In the in vitro model (sham, OGD/R, OGD/R + MS, OGD/R + Fer-1), MM cells in the MS group were cultivated with or without glucose. Fer-1 (1 μM) was used as a positive control. Finally, to determine whether MS inhibits ferroptosis via the Nrf2/HO-1 pathway, the mice were divided into four groups: the Sham group, I/R group, I/R + ML385 + MS group and I/R + MS group (n = 10, for each group). The survival rates of the mice were recorded within 48 hours (n = 15, for each group). In the I/R + ML385 + MS group, the Nrf2 inhibitor ML385 (Solarbio, Beijing, China) was administered intraperitoneally at a dose of 30 mg/kg 1 hour before the ischemia model was established. Additionally, Nrf2 was knocked out in Caco-2 cells in vitro.
Hematoxylin–eosin (H&E) staining
After 120 minutes of reperfusion, a 1 cm segment of intestinal tissue was taken 5 cm from the ileocecal valve. The intestinal tissue was fixed in 4% (w/v) paraformaldehyde and then embedded in paraffin. Sections 4 µm thick were cut for hematoxylin and eosin (H&E) staining. The histological damage of the intestinal tissue was evaluated using Chiu's scoring system, according to previously described methods [Citation8].
Transepithelial electrical resistance
To assess the integrity of the intestinal barrier after I/R, fresh intestinal mucosa was collected after the mice were euthanized. The transepithelial electrical resistance (TER) was measured using Ussing chambers (EMCSYS, Physiologic Instruments, USA) and a voltohmmeter, as previously reported [Citation20]. The calculation formula for TER is as follows: TER (Ω·cm^2) = PD/ΔIsc, as previously described [Citation20].
Western blot
Western blotting was carried out as previously described [Citation19]. The protein samples collected and homogenized from tissues and cells were quantified using the BCA protein assay for pipetting and quantification. After separation via SDS–PAGE, the protein samples were transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were then blocked with 8% (w/v) skim milk for 1 hour and incubated overnight at 4 °C with the appropriate primary antibodies: anti-GPX4 (1:1000; Proteintech, China), anti-FTH1 (1:1000; Proteintech, China), anti-Nrf2 (1:1000; Abcam, USA), and anti-HO-1 (1:1000; Abcam, USA). Subsequently, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at 37 °C for 1 hour. Protein expression was detected using a chemiluminescence (ECL) system. The reference for normalizing protein expression was β-actin.
Immunohistochemistry analysis
Immunohistochemistry (IHC) staining was conducted as previously described [Citation21]. Tissue sections were incubated with the primary antibody anti-GPX4 (1:400; Proteintech, China) at 4 °C overnight. Next, the intestinal tissues were incubated with secondary antibodies at room temperature for 1 hour to detect the expression of the GPX4 protein. The slides were stained with diaminobenzidine tetrahydrochloride (DAB), counterstained with hematoxylin, and then mounted for microscopic examination.
Immunofluorescence staining
Immunofluorescence staining was performed as previously described [Citation21]. Briefly, cells and tissue sections were washed three times with PBS containing 0.25% (v/v) Tween-20 (PBS-T), blocked with 5% goat serum, and then incubated overnight at 4 °C with primary antibodies against ZO-1 (1:200; Proteintech, China), occludin (1:200; Proteintech, China), GPX4 (1:200; Proteintech, China), FTH1 (1:200; Proteintech, China), Nrf2 (1:200; Abcam, USA), and HO-1 (1:200; Abcam, USA). Subsequently, the cells and tissue sections were incubated with fluorescent secondary antibodies (goat anti-mouse or goat anti-rabbit antibodies, Proteintech, China, diluted 1:100) for 1 hour, followed by washing with PBS. Finally, the cells and tissue sections were stained with 4'−6-diamidino-2-phenylindole (DAPI) and observed under a fluorescence microscope (Beyotime, China). Quantification was performed using ImageJ software (NIH, USA).
Cell viability assay and LDH detection
Cell viability was tested using a Cell Counting Kit-8 (CCK-8, Abcam, USA) according to the manufacturer's instructions. Cell supernatants were collected for lactate dehydrogenase (LDH) measurement following the manufacturer's instructions (Nanjing Jiancheng, China). The optical density (OD) was measured at 450 nm.
Measurements of malondialdehyde, glutathione and iron
To analyze the glutathione (GSH), malondialdehyde (MDA), and iron levels in intestinal tissues, the tissues were first homogenized in phosphate-buffered saline (PBS). The supernatant was collected after centrifugation. Then, using the appropriate assay kits and following the manufacturer's instructions, the levels of MDA (A003-1, Nanjing Jiancheng, China), GSH (BC1175, Solarbio, China), and iron (ab83366, Abcam) in the intestinal tissue supernatant were measured.
TUNEL
Transferred enzyme-mediated deoxyuridine triphosphate biotin terminal end labeling (TUNEL) staining was performed on paraffin-embedded intestinal tissue sections fixed with formaldehyde to detect apoptosis in intestinal epithelial cells. The slices were examined using a fluorescence microscope with excitation at 480 nm and emission at 530 nm. The fluorescence intensity was measured using ImageJ 2x software.
Transmission electron microscopic analysis
The intestine was incubated overnight in 2.5% (v/v) glutaraldehyde containing 0.1 M PBS (pH 7.4). The sections were then sliced into 50 µm thick sections using a vibratome. Subsequently, the sections were fixed in 1% (w/v) osmium tetroxide for 1 hour, dehydrated in 3% (w/v) uranyl acetate-ethanol solution, and embedded in epoxy resin. After polymerization, 100 nm thick slices were cut, stained with uranyl acetate and lead citrate, and examined using a transmission electron microscope. ImageJ was utilized for image analysis (NIH, USA).
Knockdown of Nrf2 in Caco-2 cells
When Caco-2 cells reached 50% – 70% confluence, Nrf2 siRNA (GeneChem, China) transfection was performed. The cells were cultured in six-well plates. After establishing the Nrf2 siRNA Caco-2 cell model, the OGD/R model was constructed.
Statistical analysis
All the data were expressed as the mean ± standard deviation (SD). The Kolmogorov–Smirnov test was employed to assess the normality of the sample distribution and conduct multiple comparisons. Statistical analyses were performed using SPSS 18.0 software (SPSS Inc., USA). Student's t test and one-way analysis of variance (ANOVA) were used for comparisons among the three groups. GraphPad (version 7) Prism software (GraphPad Software, CA) was used for all the statistical analyses. All the statistical tests were two-tailed, and a p value less than 0.05 was considered to indicate statistical significance.
Results
I/R injury induced intestinal cell damage and intestinal barrier dysfunction
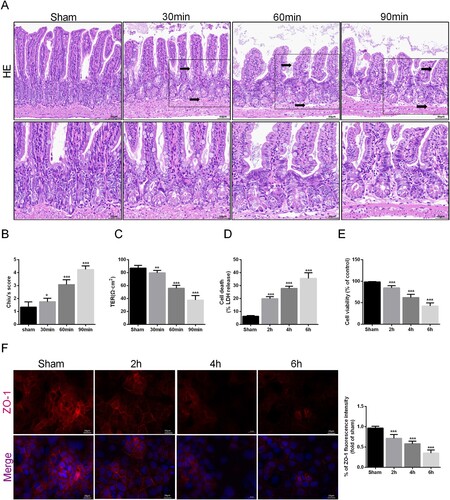
To determine the impact of intestinal I/R injury on intestinal barrier function, the mice were euthanized, and intestinal tissues were collected for HE staining and Chiu's scoring. Compared to those in the sham group, as the duration of ischemia increased, the arrangement of the intestinal epithelial tissues became progressively disordered, the villi gradually became sparse and blunted at the distal end, the gaps widened, and some villi were shed, with a gap appearing between the mucosal layer and the submucosal layer (A). The Chiu score also increased accordingly (B). The tightness of intercellular connections in intestinal epithelial cells decreased, as confirmed by the TER (C). In the in vitro experiment, we observed the release of LDH and cell viability in Caco-2 cells after OGD/R. With prolonged hypoxia, LDH release increased, and cell viability decreased, further confirming the damage to intestinal epithelial cells (D-E). In addition, the reduction in the tight junction protein ZO-1 in the in vitro caco2 cell OGD/R model indicated damage to the integrity and permeability of the intestinal mucosal barrier after hypoxia (F). Based on the abovementioned results, we proved that intestinal barrier function is damaged in a time-related way after I/R injury.
Figure 1. I/R injury induced intestinal cell damage and intestinal barrier dysfunction. Mice were subjected to I/R or sham surgery and divided into four groups: sham, 30 , 60 , and 90 min (n = 10 for each group). Intestines were collected after ischemia. (A-B) Histopathological damage was estimated with H&E staining (scale bars: 100 μm) and Chiu's score. (C) The integrity of the intestinal barrier was evaluated with TER. The OGD/R model was established in Caco-2 cells. (D) The release levels of LDH were detected. (E) Cell viability was measured with a CCK-8 assay. (F) The expression levels of ZO-1 were analyzed by IF staining (Scale bars: 20 μm). The values are shown as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the sham group.
I/R injury induced ferroptosis in the intestine
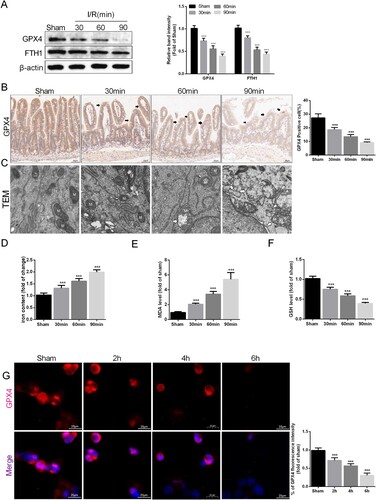
To validate whether intestinal I/R injury is related to ferroptosis, the expression of GPX4 and FTH1, two antiferroptosis-related proteins, in intestinal tissue after I/R injury was detected via Western blotting (A). The results indicated that the levels of both of these proteins in the intestinal tissue of the I/R group decreased with prolonged ischemia compared to those in the sham group. Next, we confirmed this conclusion by IHC staining of GPX4 in intestinal epithelial tissue (B). Changes in mitochondrial morphology are a major feature of ferroptosis, so we observed the morphology of mitochondria after different durations of ischemia. Compared with those in the sham group, the intestinal epithelial cell mitochondria in the I/R group exhibited clear ferroptosis characteristics, including the rupture of the mitochondrial outer membrane and the disappearance of mitochondrial cristae (C). Additionally, the level of iron ions in intestinal tissue was positively related to the I/R time (D). The levels of MDA, the final product of lipid peroxidation, were positively correlated with the duration of ischemia, while the levels of GSH, another protective factor against ferroptosis, were negatively correlated with the duration of ischemia (E-F). The in vitro IF staining results showed that GPX4 expression levels also gradually decreased with increasing ischemia duration (G). In conclusion, ferroptosis is crucial for the disruption of intestinal barrier function caused by I/R injury.
Figure 2. I/R injury induced ferroptosis in the intestine. Mice were subjected to I/R or sham surgery and divided into four groups: sham, 30 , 60 , and 90 min (n = 10 for each group). (A) The expression of GPX4 and FTH1 was measured by Western blotting. (B) The IHC staining of GPX4 was used to examine the occurrence of ferroptosis (scale bars: 50 μm). (C) Representative TEM images of mitochondrion (Scale bars: 500 nm). The levels of iron (D), GSH (E) and MDA (F) were evaluated by ELISA. The OGD/R model was established in Caco-2 cells. (G) The expression levels of GPX4 were analyzed by IF staining (Scale bars: 20 μm). The values are shown as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the sham group.
Methane saline alleviated intestinal I/R injury
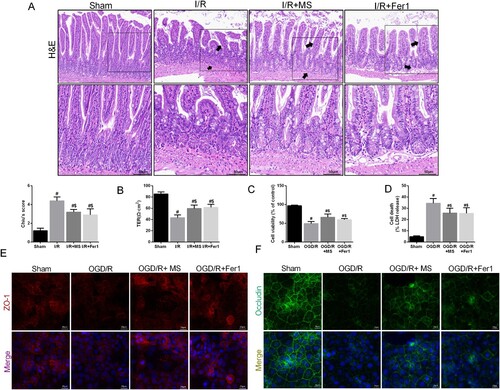
To evaluate the potential of MS to protect against intestinal I/R injury and verify the correlation between MS and ferroptosis, we administered MS intervention to mice while simultaneously inhibiting ferroptosis with Fer-1. The results of HE staining showed that the intestinal epithelial tissue damage caused by I/R injury was effectively alleviated by Fer-1 and MS treatment compared to that in the I/R group (A), which was consistent with the TER results (B). In the in vitro experiments, MS treatment effectively reduced the release of LDH in Caco-2 cells and improved cell viability (C-D). Furthermore, IF results confirmed that MS treatment could effectively increase the expression of ZO-1 and Occludin in Caco-2 cells during I/R (E-F). The results indicated that MS treatment reduced intestinal I/R injury.
Figure 3. Methane saline alleviated intestinal I/R injury. The animal models were launched with a 60-min ischemia period as mentioned. MS (10 mL/kg) was used to treat the mice as previously mentioned (n = 10 for each group). (A) Histopathological damage was estimated with H&E staining (scale bars: 100 μm) and Chiu's score. (B) The integrity of the intestinal barrier was evaluated with TER. The OGD/R model was established in Caco-2 cells. (C) The release levels of LDH were detected. (D) Cell viability was measured with a CCK-8 assay. (E-F) The expression levels of ZO-1 and Occludin were analyzed by IF staining (Scale bars: 20 μm). The values are shown as the mean ± SD. #p < 0.05, compared with the sham group, and $p < 0.05 compared with the I/R or OGD/R group.
Methane saline alleviated intestinal I/R injury by suppressing ferroptosis
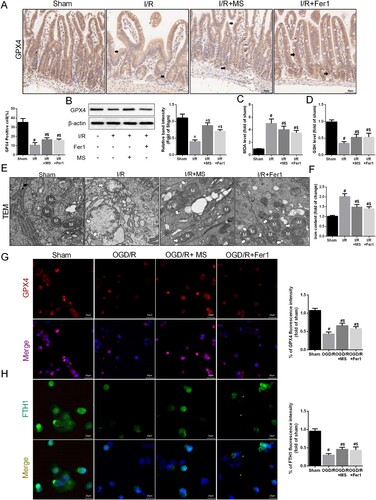
After I/R, the expression of the factor related to ferroptosis, GPX4, was suppressed. The IHC staining results indicated that MS and Fer-1 intervention effectively improved the expression of GPX4 (A). Western blot analysis also confirmed that MS and Fer-1 alleviated the inhibitory effect of I/R on GPX4 (B). MS treatment also effectively reduced the MDA levels and increased the GSH levels in the intestinal tissue during I/R injury (C-D). The TEM results also showed that MS treatment effectively improved mitochondrial damage (E). Moreover, MS treatment reduced the iron ion levels in the intestinal tissue during I/R injury (F). We also assessed the effect of MS on GPX4 and FTH1 expression in Caco-2 cells by using IF staining (G-H). The results showed that MS therapy effectively promoted GPX4 and FTH1 expression and reduced intestinal ferroptosis in Caco-2 cells during I/R injury. In summary, MS treatment improves intestinal barrier function by inhibiting ferroptosis in intestinal epithelial cells during I/R injury.
Figure 4. Methane saline alleviated intestinal I/R injury via suppressing ferroptosis. The animal models were launched with a 60-min ischemia period as mentioned. MS (10 mL/kg) was used to treat the mice as previously mentioned (n = 10 for each group). (A) Immunohistochemical staining of GPX4. (B) The expression of GPX4 was measured by Western blotting. The levels of MDA (C) and GSH (D) were evaluated by ELISA. (E) Representative TEM images of mitochondrion (Scale bars: 500 nm). (F) The levels of iron were evaluated by ELISA. The OGD/R model was established in Caco-2 cells. (G-H) The expression levels of GPX4 and FTH1 were analyzed by IF staining (Scale bars: 20 μm). The values are shown as the mean ± SD. #p < 0.05, compared with the sham group, and $p < 0.05 compared with the I/R or OGD/R group.
Methane saline suppresses ferroptosis in intestinal I/R injury through the Nrf2/HO-1 signaling pathway
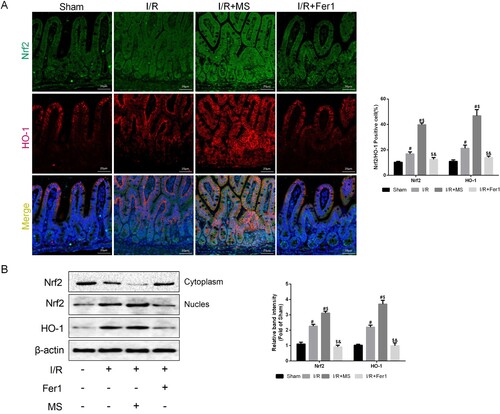
Research has demonstrated that Nrf2/HO-1 can effectively improve ferroptosis in cerebral I/R injury [Citation22]. MS has been shown to promote Nrf2 expression and alleviate acetylaminophen-induced liver damage [Citation18]. Therefore, we evaluated Nrf2/HO-1 expression in intestinal tissues using IF staining (A). MS treatment effectively promoted the expression of Nrf2 (green) and HO-1 (red) compared to that in the I/R group, while Nrf2/HO-1 expression was not affected by Fer-1 intervention. WB results also confirmed this conclusion (B). Therefore, we believe that MS may alleviate ferroptosis in intestinal epithelial cells during I/R through enhancing the Nrf2/HO-1 signaling pathway.
Figure 5. Methane saline promotes the expression of Nrf2/HO-1 signaling Pathway. The animal models were launched with a 60-min ischemia period as mentioned. MS (10 mL/kg) was used to treat the mice as previously mentioned (n = 10 for each group). (A) he colocalization of Nrf2 and HO-1 was shown by immunofluorescence (Scale bars: 50 μm). (B) The expression of Nrf2 and HO-1 was measured by Western blotting. The values are shown as the mean ± SD. #p < 0.05, compared with the sham group, and $p < 0.05 compared with the I/R group, &p < 0.05 compared with the I/R + MS group.
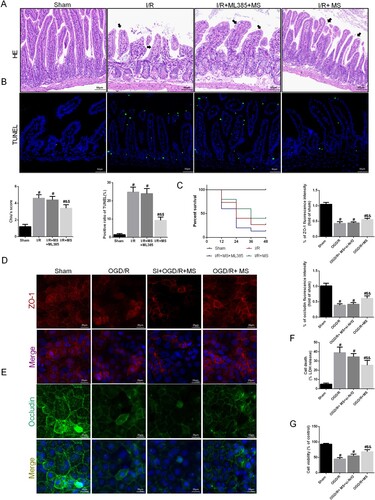
To further confirm whether MS activates the Nrf2/HO-1 signaling pathway to alleviate ferroptosis during I/R injury, we inhibited the expression of Nrf2 in mice using ML385 and simultaneously administered MS intervention to verify the correlation between MS and Nrf2. We used the immunofluorescence staining of Nrf2 and HO-1 in intestinal tissue to demonstrate the inhibition effect of ML385 (Fig. S1 A). The HE staining results showed that the ability of MS to protect intestinal epithelial cells and the intestinal barrier was suppressed after Nrf2 was inhibited. This was reflected by worsened HE staining damage and decreased Chius scores (A). The TUNEL staining results was consistent with the HE staining (B). We also found that mice treated with MS had a greater survival rate after intestinal I/R injury, but this protective effect was inhibited after Nrf2 was inhibited (C). In the in vitro experiments, we further silenced Nrf2 in Caco-2 cells to explore the protective effect of MS on intestinal I/R injury in a Nrf2-dependent manner. We used the immunofluorescence staining of Nrf2 and HO-1 to evaluate the knockdown efficiency of Nrf2 si-RNA (Fig. S1 B). The IF staining results showed that inhibiting Nrf2 reduced the protective impact of MS on intestinal barrier function, resulting in decreased expression of the intercellular tight junction proteins ZO-1 and Occludin (D-E). Cell viability and LDH levels also verified these results (F-G).
Figure 6. Knockdown of Nrf2 abolished MS-alleviated intestinal I/R injury. The animal models were launched with a 60-min ischemia period as mentioned. MS (10 mL/kg) was used to treat the mice as previously mentioned and ML385, a specific inhibitor of Nrf2, was used as a positive control (n = 10 for each group). (A) Histopathological damage was estimated with H&E staining (scale bars: 100 μm) and Chiu's score. (B) Survival rates were calculated in different groups (n = 15). (C) Representative images of TUNEL staining of intestinal sections (Scale bars: 50 μm). (D) The integrity of the intestinal barrier was evaluated with serum I-FABP levels. Caco-2 cells were transfected with Nrf2-targeting siRNA. Then the OGD/R model was established in Caco-2 cells. (E-F) The expression levels of ZO-1 and Occludin were analyzed by IF staining (Scale bars: 20 μm). (G) The release levels of LDH were detected. (H) Cell viability was measured with a CCK-8 assay. The values are shown as the mean ± SD. #p < 0.05, compared with the sham group, and $p < 0.05 compared with the I/R or OGD/R group, &p < 0.05 compared with the I/R + MS + Nrf2 – / – group.
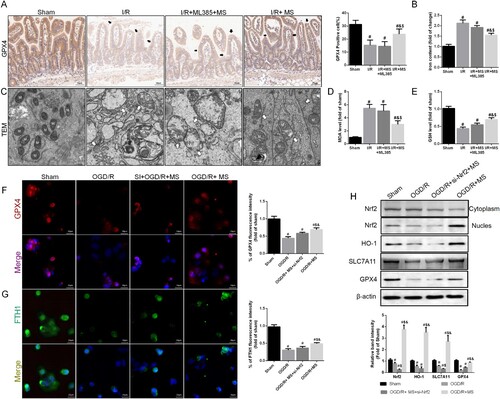
Finally, we assessed the impact of Nrf2 inhibition on the effect of MS on intestinal I/R injury. IHC results for GPX4 indicated that MS could effectively increase the expression of GPX4 during I/R injury, and this effect was inhibited after silencing Nrf2 (A). We then measured the iron ion levels in intestinal tissues, and the effect of MS on reducing iron ion levels was also reversed after inhibiting Nrf2 (B). TEM results also suggested that after Nrf2 was silenced, the protective effect of MS on mitochondria was inhibited (C). Furthermore, the inhibition of Nrf2 weakened the antioxidant effect of MS, as reflected by increased MDA levels and decreased GSH levels in the intestinal tissues (D-E). In vitro IF staining also confirmed that the absence of Nrf2 reversed the inhibitory effect of MS on ferroptosis, as indicated by the reduced expression of GPX4 and FTH1 in Caco-2 cells (F-G). We used western blot to measured the expression of Nrf2 and its downstream proteins (H). The results shown that MS treatment could effectively improve the expression of HO-1and SLC7A11, silencing the expression of Nrf2 reversed this results. Meanwhile, inhibiting the expression of Nrf2 also reduced the activity of GPX4. In summary, MS suppresses intestinal I/R injury-induced ferroptosis by activating the Nrf2/HO-1 signaling pathway, thereby alleviating intestinal barrier damage.
Figure 7. Knockdown of Nrf2 abolished MS-inhibited ferroptosis. The animal models were launched with a 60-min ischemia period as mentioned. MS (10 mL/kg) was used to treat the mice as previously mentioned and ML385, a specific inhibitor of Nrf2, was used as a positive control (n = 10 for each group). (A) Immunohistochemical staining of GPX4 (Scale bars: 50 μm). (B) the levels of iron were evaluated by ELISA. (C) Representative TEM images of mitochondrion (Scale bars: 500 nm). The levels of MDA (D) and GSH (E) were evaluated by ELISA. Caco-2 cells were transfected with Nrf2-targeting siRNA. Then the OGD/R model was established in Caco-2 cells. (F-G) The expression levels of GPX4 and FTH1 were analyzed by IF staining (Scale bars: 20 μm). (H) The expression of Nrf2, HO-1 and GPX4 was measured by Western blotting. The values are shown as the mean ± SD. #p < 0.05, compared with the sham group, and $p < 0.05 compared with the I/R group.
Discussion
Intestinal ischemia–reperfusion injury is a significant problem for surgeons due to its high incidence rate. Reactive oxygen species are considered one of the most important factors in intestinal I/R injury. Our prior studies demonstrated that MS possesses antioxidative properties that can reduce sepsis-induced liver injury [Citation17]. However, studies on the effects of MS on intestinal IR injury are still scarce. Our study revealed that I/R injury can cause damage to intestinal barrier function over time, and this damage is also closely associated with ferroptosis. After confirming that ferroptosis is involved in intestinal I/R injury, we verified the protective effects of MS on intestinal I/R injury. Initially, we found that MS treatment could alleviate damage to intestinal epithelial cells and the intestinal barrier. Fer-1 can form complexes with ferrous ions and eliminate alkoxyl radicals produced by ferrous ions, thereby mitigating ferroptosis [Citation21]. We next examined the influence of MS on ferroptosis. We found that MS treatment can increase the levels of GPX4 and FTH1, which are crucial regulators of ferroptosis, both in vivo and in vitro during I/R injury. Moreover, we detected that MS treatment increased the Nrf2 and HO-1 levels in intestinal epithelial cells, suggesting that MS inhibits ferroptosis through the Nrf2/HO-1 pathway to alleviate intestinal I/R injury. After Nrf2 was knocked out, both the protective effect of MS on the intestine and its inhibition of ferroptosis were reversed, further confirming our findings.
Ferroptosis is a recently discovered type of programable cell death that is considered to be dependent on high levels of iron and the formation of ROS. It has been found to be closely related to I/R injury [Citation23]. Glutathione is used as a coenzyme by GPX4, a biomarker of ferroptosis. Thus, ferroptosis can be caused by binding to and inhibiting GPX4 or by reducing the amount of glutathione available within the cell.
The depletion of GSH decreases the expression of GPX4, a crucial process that occurs upstream of mitochondrial dysfunction, leading to mitochondrial dysfunction, which in turn causes ROS to accumulate and ultimately causes ferroptosis [Citation24, Citation25]. Moreover, the vital mechanism of ferroptosis, lipid peroxidation, has been demonstrated to be important for intestinal I/R injury [Citation26]. Our research demonstrated a decrease in the expression of the antioxidant factors GPX4 and GSH and that the expression of the lipid peroxidation product MDA increased during intestinal I/R injury in a time-dependent manner. Nevertheless, MS treatment may be able to partially reverse these consequences. Thus, we believe that MS can inhibit ferroptosis to ameliorate intestinal I/R injury.
Due to its tiny molecule size and distribution, which allow it to cross membranes and diffuse into organelles, methane has been suggested as a potential treatment gas for ischemia/reperfusion injury [Citation27]. However, the effect of methane on I/R injury-induced ferroptosis remains unknown. Our research revealed that MS appropriately protected the intestinal barrier from I/R injury by decreasing damage to tight junction proteins, preserving the structure of the intestinal epithelial tissue and reducing ferroptosis. Notably, MS treatment also improved the expression of Nrf2 and HO-1. A study revealed that Nrf2 could mitigate acute lung injury during I/R injury by regulating ferroptosis via the expression of SLC7A11 and HO-1 [Citation28]. MS can prevent liver injury induced by acetaminophen (APAP) through anti-inflammatory, antioxidant, anti-ER stress (ER), and antiapoptotic effects mediated through the Nrf2/HO-1/NQO1 signaling pathway [Citation18]. Based on previous research, we attempted to link the protective effects of MS on intestinal I/R injury with ferroptosis and the Nrf2/HO-1 signaling pathway.
NRF2 is a transcription factor activated by stress. Under physiological conditions, the cytoplasmic protein Kelch-like ECH-associated protein 1 (Keap1) binds to NRF2, inhibiting NRF2 activation by mediating its ubiquitination and degradation. When exposed to oxidative or electrophilic stress, cysteine residues within the Keap1 structural domain are covalently modified, leading to decreased stability of Keap1-NRF2 binding, thereby releasing NRF2 from inhibition. As a result, NRF2 is released from Keap1, enters the nucleus, and recognizes the antioxidant response element (ARE), thereby inducing the expression of genes encoding antioxidative stress-related enzymes [Citation29]. Currently, Nrf2 has been shown to exert antiferroptotic effects by regulating oxidative stress, iron, and lipid metabolism [Citation30].
The metabolic enzyme heme oxygenase-1 (HO-1) can be stimulated by heme and various stresses, such as hydrogen peroxide, hypoxia, and inflammation, and is a core participant in the mammalian stress response. Current evidence suggests that HO-1 primarily protects against intestinal I/R injury by metabolizing toxic heme into carbon monoxide (CO), Fe2+, and biliverdin [Citation31]. Therefore, proper NRF2 and HO-1 function is crucial for cell survival, especially during periods of increased oxidative stress. Recent research has confirmed that NRF2 can regulate the activation of HO-1 [Citation32]. Although HO-1 has been proven to be crucial for preventing oxidative stress, its role in ferroptosis remains controversial. Therefore, the relationship between HO-1 and ferroptosis in intestinal I/R still needs further investigation.
It is commonly believed that biological methane is produced by the anaerobic decomposition of organic matter by methanogenic archaea, but recent studies have shown that other organisms, including human cell lines, can release methane under conditions of oxidative stress triggered by free iron or reactive oxygen species without the need for specific enzymes in the presence of methyl donors [Citation33]. This finding suggests that the cells themselves can produce and utilize methane as a signaling molecule to respond to oxidative stress, providing a further theoretical basis for the use of methane in treating I/R injury. Previously, it was speculated that methane could protect cells by altering the physicochemical properties of certain proteins within cells or affecting signal transduction in the cell membrane [Citation34]. According to our research, after MS treatment, the level of Fe2 + in intestinal epithelial cells decreases, and the level of FTH1 in Caco-2 cells increases. Next, to determine whether the Nrf2/HO-1 signaling pathway mediates the mitigating effect of MS on ferroptosis, we used ML385 and siRNA to knockdown the expression of Nrf2. This effect was reversed after Nrf2 knockout, suggesting that MS can influence iron metabolism through Nrf2. Similarly, Nrf2 knockout reversed the effects of MS on increasing GPX4 and GSH levels, increasing MDA levels, and further exacerbating mitochondrial damage, further suggesting that ferroptosis inhibition by MS is partly achieved through Nrf2. Additionally, we observed that in the fer-1 treatment group, the levels of NRF2 and HO-1 decreased, which could be correlated with fer-1 reducing Fe2+-associated oxidative stress and decreasing the stimulation of NRF2 and HO-1, thus preventing ferroptosis. MS treatment causes an increase in Nrf2 and HO-1 under I/R conditions, indicating that MS might have an important function as a signaling molecule upstream of ferroptosis in the induction of Nrf2 and HO-1. Whether this effect occurs at the protein level or at the genetic level requires further proof. In summary, we believe that MS increases the expression of the Nrf2/HO-1 signaling pathway in the intestine, thereby enhancing its ‘reserve capacity’ to counteract ferroptosis during subsequent reperfusion.
Conclusion
In conclusion, we proved that intestinal I/R injury was mitigated by MS and that the underlying mechanism involved modulating the Nrf2/HO-1 signaling pathway to decrease ferroptosis. MS could be a promising treatment for intestinal I/R injury. However, there are still some limitations that must be addressed. Confirming the treatment impact of MS will require more prospective clinical trials. The results of our study indicate that MS may have significant therapeutic potential for alleviating intestinal ischemia–reperfusion injury.
Authors’ contributions
Qingrui Fan participated in designing the study and assisting in the division of labor, writing and revising the paper; Hulin Chang and Lifei Tian participated in data arrangement; Bobo Zheng and Ruiting Liu participated in revision; Zeyu Li participated in designing the study and assisting in the division of labor, writing and revising the paper.
Ethics approval and consent to participate
This study was approved and carried out by the ethics committee of Shaanxi Provincial People's Hospital and strictly adhered to the tenets of the Declaration of Helsinki code of Ethical approval for scientific research project: 2019 Ethical Scientific Research Approva.
Acknowledgments
We are indebted to all individuals who participated in or helped with this research project.
Availability of data and materials
The data used to support the findings of this study are included in the paper.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Zhu T, Wan Q. Pharmacological properties and mechanisms of Notoginsenoside R1 in ischemia-reperfusion injury. Chin J Traumatol. 2023;26:20–26. doi:10.1016/j.cjtee.2022.06.008
- Gonzalez LM, Moeser AJ, Blikslager AT. Animal models of ischemia-reperfusion-induced intestinal injury: progress and promise for translational research. Am J Physiol Gastrointest Liver Physiol. 2015;308:G63–G75. doi:10.1152/ajpgi.00112.2013
- Zhou L, Han S, Guo J, et al. Ferroptosis-a new dawn in the treatment of organ ischemia-reperfusion injury. Cells. 2022;11(22):3653. doi:10.3390/cells11223653
- Chen F, Liu Y, Shi Y, et al. The emerging role of neutrophilic extracellular traps in intestinal disease. Gut Pathog. 2022;14:27, doi:10.1186/s13099-022-00497-x
- Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi:10.1016/j.cell.2012.03.042
- Agmon E, Solon J, Bassereau P, et al. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep. 2018;8:5155, doi:10.1038/s41598-018-23408-0
- Li Y, Feng D, Wang Z, et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019;26:2284–2299. doi:10.1038/s41418-019-0299-4
- Feng YD, Ye W, Tian W, et al. Old targets, new strategy: apigenin-7-O-β-d-(-6″-p-coumaroyl)-glucopyranoside prevents endothelial ferroptosis and alleviates intestinal ischemia-reperfusion injury through HO-1 and MAO-B inhibition. Free Radic Biol Med. 2022;184:74–88. doi:10.1016/j.freeradbiomed.2022.03.033
- Poles MZ, Juhasz L, Boros M. Methane and inflammation – a review (fight fire with fire). Intensive Care Med Exp. 2019;7:68, doi:10.1186/s40635-019-0278-6
- Keppler F, Boros M, Polag D. Radical-driven methane formation in humans evidenced by exogenous isotope-labeled DMSO and methionine. Antioxidants (Basel). 2023;12(7):1381. doi:10.3390/antiox12071381
- Chen O, Ye Z, Cao Z, et al. Methane attenuates myocardial ischemia injury in rats through anti-oxidative, anti-apoptotic and anti-inflammatory actions. Free Radic Biol Med. 2016;90:1–11. doi:10.1016/j.freeradbiomed.2015.11.017
- Long C, Yang J, Yang H, et al. Attenuation of renal ischemia/reperfusion injury by oleanolic acid preconditioning via its antioxidant, anti-inflammatory, and anti-apoptotic activities. Mol Med Rep. 2016;13:4697–4704. doi:10.3892/mmr.2016.5128
- Ye ZH, Ning K, Ander BP, et al. Therapeutic effect of methane and its mechanism in disease treatment. J Zhejiang Univ Sci B. 2020;21:593–602.
- Boros M, Ghyczy M, Erces D, et al. The anti-inflammatory effects of methane. Crit Care Med. 2012;40:1269–1278. doi:10.1097/CCM.0b013e31823dae05
- Meszaros AT, Buki T, Fazekas B, et al. Inhalation of methane preserves the epithelial barrier during ischemia and reperfusion in the rat small intestine. Surgery. 2017;161:1696–1709. doi:10.1016/j.surg.2016.12.040
- Li Z, Jia Y, Feng Y, et al. Methane alleviates sepsis-induced injury by inhibiting pyroptosis and apoptosis: in vivo and in vitro experiments. Aging (Albany NY). 2019;11:1226–1239. doi:10.18632/aging.101831
- Li Z, Jia Y, Feng Y, et al. Methane-rich saline protects against sepsis-induced liver damage by regulating the PPAR-γ/NF-κB signaling pathway. Shock. 2019;52:e163–e172. doi:10.1097/SHK.0000000000001310
- Feng Y, Cui R, Li Z, et al. Methane alleviates acetaminophen-induced liver injury by inhibiting inflammation, oxidative stress, endoplasmic reticulum stress, and apoptosis through the Nrf2/HO-1/NQO1 signaling pathway. Oxid Med Cell Longev. 2019;2019:7067619.
- Li Z, Wang B, Tian L, et al. Methane-rich saline suppresses ER-mitochondria contact and activation of the NLRP3 inflammasome by regulating the PERK signaling pathway to ameliorate intestinal ischemia–reperfusion injury. Inflammation. 2024;47:376–389.
- Cheng J, Zhang L, Dai W, et al. Ghrelin ameliorates intestinal barrier dysfunction in experimental colitis by inhibiting the activation of nuclear factor-kappa B. Biochem Biophys Res Commun. 2015;458:140–147. doi:10.1016/j.bbrc.2015.01.083
- Miotto G, Rossetto M, Di Paolo ML, et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020;28:101328.
- Li J, Wei G, Song Z, et al. SIRT5 regulates ferroptosis through the Nrf2/HO-1 signaling axis to participate in ischemia-reperfusion injury in ischemic stroke. Neurochem Res. 2024;28: 101328. doi:10.1016/j.redox.2019.101328
- Zhu K, Fan R, Cao Y, et al. Glycyrrhizin attenuates myocardial ischemia reperfusion injury by suppressing Inflammation, oxidative stress, and ferroptosis via the HMGB1-TLR4-GPX4 pathway. Exp Cell Res. 2024;435:113912, doi:10.1016/j.yexcr.2024.113912
- Zhang W, Gong M, Zhang W, et al. Thiostrepton induces ferroptosis in pancreatic cancer cells through STAT3/GPX4 signalling. Cell Death Dis. 2022;13:630, doi:10.1038/s41419-022-05082-3
- Zhu K, Zhu X, Sun S, et al. Inhibition of TLR4 prevents hippocampal hypoxic-ischemic injury by regulating ferroptosis in neonatal rats. Exp Neurol. 2021;345:113828, doi:10.1016/j.expneurol.2021.113828
- Zhang J, Song L, Xu L, et al. Knowledge domain and emerging trends in ferroptosis research: a bibliometric and knowledge-map analysis. Front Oncol. 2021;11:686726, doi:10.3389/fonc.2021.686726
- Zhang B, Tian X, Li G, et al. Methane inhalation protects against lung ischemia-reperfusion injury in rats by regulating pulmonary surfactant via the Nrf2 pathway. Front Physiol. 2021;12:615974. doi:10.3389/fphys.2021.615974
- Dong H, Qiang Z, Chai D, et al. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging (Albany NY). 2020;12:12943–12959. doi:10.18632/aging.103378
- Baird L, Yamamoto M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol Cell Biol. 2020;40.
- Shakya A, McKee NW, Dodson M, et al. Anti-ferroptotic effects of Nrf2: beyond the antioxidant response. Mol Cells. 2023;46:165–175.
- Liao YF, Zhu W, Li DP, et al. Heme oxygenase-1 and gut ischemia/reperfusion injury: a short review. World J Gastroenterol. 2013;19:3555–3561. doi:10.3748/wjg.v19.i23.3555
- Alam J, Stewart D, Touchard C, et al. Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274:26071–8. doi:10.1074/jbc.274.37.26071
- Boros M, Keppler F. Methane production and bioactivity-a link to oxido-reductive stress. Front Physiol. 2019;10:1244, doi:10.3389/fphys.2019.01244
- Wang W, Huang X, Li J, et al. Methane suppresses microglial activation related to oxidative, inflammatory, and apoptotic injury during spinal cord injury in rats. Oxid Med Cell Longev. 2017: 2190897. doi:10.1155/2017/2190897