
1. Introduction
The search for the magic bullet in cancer immunotherapies is ongoing since more than 100 years, but only recent clinical success of immune checkpoint directed antibodies significantly revived the field of immune-oncology [Citation1]. A key protein target in this area is the protein–protein interaction (PPI) between PD-1 and its ligand PD-L1. Functionally, PD-1, also called programmed death-1 protein, comprises an immune checkpoint located on T-cells. The PD-1/PD-L1 axis is hijacked by viruses and tumor/cancer cells to suppress the immune surveillance. For example, PD-L1 is expressed on tumor cells and also on immune cells (e.g. myeloid tumor-infiltrating cells). Binding of PD-1 to PD-L1 determines a downregulation of T-cell effector functions in cancer patients, inhibiting the antitumor immune response and leading to T-cell exhaustion [Citation2]. In viral diseases, a similar mechanism is used by viruses to undermine the effective immune recognitions [Citation3]. Current medication directed toward the PD-1/PD-L1 axis includes monoclonal antibodies. These have shown impressive clinical results in the treatment of several types of tumors, including melanoma and lung cancer [Citation4]. Currently, two humanized monoclonal antibodies targeting PD-1 are approved by the regulatory bodies, pembrolizumab and nivolumab [Citation5]. Multiple additional clinical trials either as single agents or in combination with other agents are ongoing to extend their indication areas. Therapeutic antibodies however exhibit several disadvantages such as limited tissue and tumor penetration, very long half-life time, lacking oral bioavailability, immunogenicity, and difficult and expensive production. Moreover, current PD-1/PD-L1 axis directed monoclonal antibodies lead to a tumor response only in a fraction of cases and tumor types. Therefore, a search for non-mAbs, including small molecules, peptides, cyclo-peptides, and macrocycles is ongoing and will be reviewed here.
2. Body
Recently the co-crystal structure between the human PD-1 and PD-L1 has been determined for the first time [Citation6]. The Å-resolution crystal structure provides a possible starting point for the design of molecules against the PPI. The interface between the two proteins is extended (~1.700 Å2), hydrophobic and flat, without deep binding pockets, which makes the interface likely a difficult target for small molecules. The hydrophobic interface also increases the chances to discover false positive hits considerably. Moreover, direct competitor PD-1/PD-L1 antagonists are potentially very hydrophobic molecules, which can lead to downstream development issues, including toxicity, selectivity, and poor water solubility, just to name a few. Nonetheless, currently several series of small molecules, peptides, and cyclo-peptides have been disclosed targeting the PD-1/PD-L1 PPI. In the following, we focus on discussing first small molecules, followed by peptide and cyclic peptide derivatives.
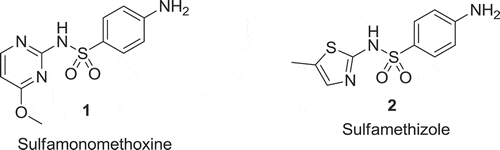
A group from Harvard University has discovered sulphonamide derivatives (1) and (2) to work in a similar fashion as reference mAbs [Citation7] (). The two compounds are active antagonists in an IFNγ-release assay in transgenic mouse T-cells that express PD-1.
Figure 1. Small-molecule antagonists of the PD-1 pathway.
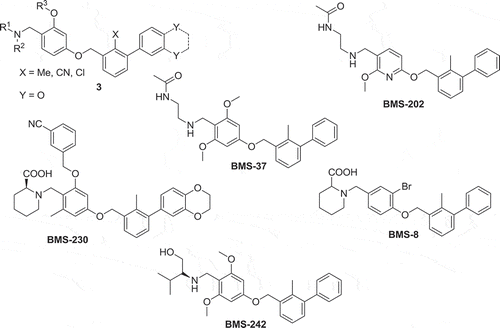
Workers from the company Bristol-Myers Squibb (BMS) have disclosed scaffold (3) () binding to PD-L1 [Citation8]. It consists of a tri-aromatic structure, including a mono-ortho substituted biphenyl substructure. Moreover, another phenyl ring is connected to the biphenyl and contains also a methylene amine moiety. The biological activity of the claimed compounds was established by a homogenous time-resolved fluorescence (HTRF) binding assay in which Europium cryptate-labeled anti-Ig was used. To assess selectivity, the PPIs PD-1/PD-L2 and PD-L1/CD80 were tested as well. Typical examples are BMS-8, BMS-37, BMS-202, BMS-230, and BMS-242. No further in vitro or in vivo assays have been described supporting the biological activity of compounds based on scaffold (3) (). The structural basis of BMS-202 and BMS-8 as PD-1/PD-L1 antagonists was recently rigorously proven by a co-crystal structure and other biophysical methods [Citation9].
Figure 2. PD-1/PD-L1 inhibitors synthesized by BMS.
Certain compound classes have been recently described to interfere with the time-resolved fluorescence resonance energy transfer (TR-FRET) for the PD-1/PD-L1 assay during a high-throughput screening campaign, producing false positive hits [Citation10]. Examples include the salicylates NCI 211717 and NCI 211845. Mechanistically, the interaction of the chelator moiety salicylate with the cryptand-ligated europium FRET donor leading to a change in the assay signal is suggested ().
Figure 3. False positive PD-1/PD-L1 inhibitors.
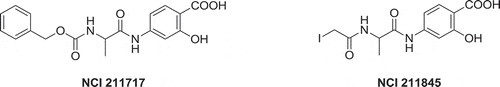
Several PD-1/PD-L1 antagonists based on peptide structures including bioisosteres such as oxadiazole and urea have been disclosed. Workers from Aurigene Ltd. described cyclic peptidomimetic compounds as immunomodulators able to interfere with the programmed death (PD-1) signaling pathway. The general formula (4) is exemplified in 20 explicit examples. It consists of a central 1-oxa-, or 1-thia-3,4-diazole fragment with a serine or threonine side chain in Position 2 and another amino acid or dipeptide linked in Position 5. Dipeptides are bound by a urea unit. In the majority of examples the first aminoacid in Position 5 is asparagine, glutamine, aspartic acid, or glutamic acid. The biological activity of the compounds was tested by using a rescue assay of mouse splenocytes in the presence of recombinant mouse PD-1/PD-L1. Compound (5) for example was able to rescue the mouse immune cells to 92% at 100 nM ().
Figure 4. Peptidomimetic inhibitors of the PD-1/PD-L1 interaction with oxa- and thiadiazole core moieties.
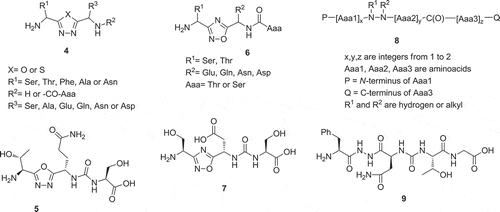
Positional isomeric 1-oxa-2,4-diazoles (6) with otherwise similar sidechains were also disclosed by the same company. Compound (7) exhibited 91% rescue in the above mouse splenocyte assay ().
Aurigene Ltd. described also small peptidomimetics comprising of 3–4 amino acid moieties comprising a hydrazine and a urea linker with general structure (8). A total of 14 compounds were explicitly exemplified [Citation11]. Compound (9) for example was able to rescue the mouse immune cells to 84% at 100 nM ().
A team from Zhengzhou and Tsinghua Universities described the discovery of the first hydrolysis-resistant D-peptide antagonists to target the PD-1/PD-L1 interaction [Citation12]. The optimized compound DPPA-1 could bind PD-L1 at an affinity of 0.51 μM in vitro. A blockade assay at the cellular level and tumor-bearing mice experiments indicated that DPPA-1 could also effectively disrupt the PD-1/PD-L1 interaction in vivo ().
Figure 5. a) Hydrolysis-resistant D-peptide antagonist to target the PD-1/PD-L1 b) Macrocyclic peptidic inhibitor c) Peptide antagonist of PD-L1 (10) along with its sequence similarities to PD-1.
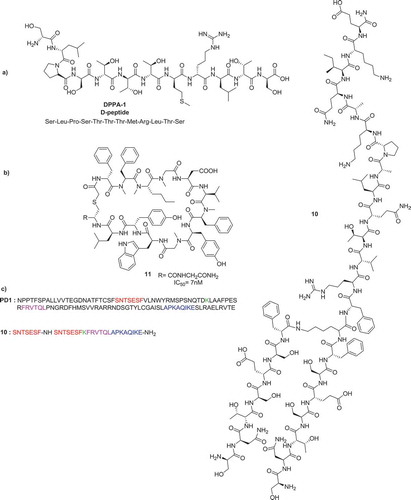
Researchers at Aurigene Ltd. developed compound (10) for the treatment of cancer. Sequences of the extracellular domain of PD-1 critical for the PD-L1/PD-L2 binding interaction, overlapping or in close proximity to the known ligand-binding regions, were identified and used as starting points for the design and evaluation of 7- to 30-mer peptides derived from human and murine PD-1 sequences. Compound (10) is highly effective in antagonizing PD-1 signaling, with in vivo exposure upon subcutaneous dosing. It is claimed to inhibit tumor growth and metastasis in preclinical models of cancer and to be well tolerated with no obvious toxicity at any of the tested doses. The structure of compound (10) is shown in [Citation13].
BMS chemists disclosed macrocyclic peptides with general structure (11) that inhibit the PD-1/PD-L1 with nanomolar potency in HTRF assays, as well as cellular-binding assays () [Citation8]. In addition, the peptides also demonstrate biological activity in cytomegalovirus (CMV) recall and HIV Elispot assays demonstrating their utility in ameliorating hyperproliferative disorders such as cancer. The peptides are competing with the binding of PD-L1 with anti-PD-1 monoclonal antibody nivolumab (BMS-936558, MDX-1106) that are known to block the interaction with PD-1, enhancing CMV-specific T-cell IFNγ secretion and enhancement of HIV-specific T-cell IFNγ secretion.
3. Expert opinion
Small molecules and peptide antagonists of the PD-1/PD-L1 interaction are highly sought after, since they could have considerable therapeutic advantages over the current clinically used mAbs. The PD-1/PD-L1 target consists of a very hydrophobic, large and flat PPI and would be classically described as ‘undruggable’ by small molecules. Nonetheless, several compound classes have been described as PD-1/PD-L1 antagonists. Currently, the majority of compound classes seem to competitively antagonize the PD-1/PD-L1 interaction and no allosteric mechanism was established. It should be mentioned that care has to be taken during the screening process to avoid false positives by carefully triaging compound hits. The sequence and structural differences between mouse and human PD-1/PD-L1 should be taken into account when recombinant proteins or mouse models are used to screen compounds. The hydrophobicity and large molecular weight of direct PD-1/PD-L1 antagonists might also result in development issues such as low target selectivity, poor oral bioavailability, low solubility, fast metabolism, and toxicity. For example, many compounds based on scaffold (3) have rather high cLogP. The nature of the PD-1/PD-L1 interaction has also resulted into several distinct peptides, including linear and cyclic peptides derived from interfacial epitopes or discovered by other techniques. Small molecule PD-1/PD-L1 antagonists might be advantageous over current mAbs due to their easy diffusion across physiologic barriers such as the blood–brain barrier and plasma membranes resulting in an overall better tumor tissue uptake. In contrast, large biomolecules such as mAbs largely rely on tumor endothelium permeability to penetrate solid tumors, and endocytic consumption can strongly affect their biodistribution. A recently described PD-1-targeting high affinity small protein, 100× smaller than a mAb and lacking its Fc moiety, penetrates deeper into tumors as seen by fluorescence microscopy and, unlike antibodies, does not cause unwanted depletion of PD-L1–positive T-cells that mediate antitumor immunity [Citation14]. PD-1/PD-L1 antagonists are proven useful in the indication areas cancer and potentially useful in viral or bacterial infections and most recently therapeutic application in Alzheimers disease (AD) are also claimed. In the first area, mAbs are well established as therapeutic agents, not so in the field of viral and bacterial infections. Small molecules and peptides antagonizing PD-1/PD-L1 might therefore easily penetrate the later indications. Moreover, in AD small molecules might be advantageous due to their blood–brain penetration. The area of non-mAb-based PD-1/PD-L1 targeting is at the very beginning and currently none of the discussed compound classes is reported to have progressed further to clinical trials yet. Thus it remains to be seen if the protein- (and cell-) based activities translate in vivo and lead to an advancement in cancer treatment and/or in the anti-infectiveor AD areas in clinical trials. Advanced techniques such as radiotracers for immunoPETimaging of PD-1 checkpoint expression on tumor-infiltrating lymphocytes and PD-L1 in general might therefore become very valuable to assess the prognostic value of PD-1/PD-L1 axis targeting molecules in preclinical models of immunotherapy and may ultimately aid in predicting response to therapies targeting immune checkpoints [Citation15].
Declaration of interest
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Dömling A, Holak TA. Programmed death-1: therapeutic success after more than 100 years of cancer immunotherapy. Angew Chem Int Ed. 2014;53:2286–2288.
- Keir ME, Butte MJ, Freeman GJ, et al. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704.
- Day CL, Kaufmann DE, Kiepiela P, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354.
- Carvalho S, Levi-Schaffer F, Sela M, et al. Immunotherapy of cancer: from monoclonal to oligoclonal cocktails of anti-cancer antibodies: IUPHAR review X. Br J Pharmacol. 2016;173:1407–1424.
- Wolchok JD. PD-1 blockers. Cell. 2015;162:937.
- Zak KM, Radoslaw K, Sara P, et al. Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure. 2015;23:2341–2348.
- President and Fellows of Harvard College. Modulators of immunoinhibitory receptor PD-1, and methods of use thereof. WO2011/082400 A3 2011.
- Bristol-Myers Squibb Company. Compounds useful as immunomodulators. WO2015/034820 A1. 2015.
- Zak KM, Grudnik P, Guzik K, et al. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget. 2016;7:30323–30335.
- Hanley RP, Horvath S, An J, et al. Salicylates are interference compounds in TR-FRET assays. Bioorg Med Chem Lett. 2016;26:973–977.
- Aurigene Discovery Technologies Limited. 1,3,4-oxadiazole and 1,3,4-thiadiazole derivatives as immunomodulators. WO2015/033301 A1. 2015.
- Chang H-N, Liu B-Y, Qi Y-K, et al. Blocking of the PD-1/PD-L1 interaction by a D-peptide antagonist for cancer immunotherapy. Angew Chem Int Ed. 2015;54:11760–11764.
- Aurigene Discovery Technologies Limited. Immunosuppression modulating compounds. US2011/0318373 A3. 2011.
- Mautea RL, Gordona SR, Mayere AT, et al. Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging. PNAS. 2015;112:6506–6514.
- Baruch K, Deczkowska A, Rosenzweig N, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat Med. 2016;22:135–137.