
ABSTRACT
Introduction: Diffuse large B cell lymphoma (DLBCL) is the most frequent lymphoma in adults. 30–40% DLBCL eventually relapse and 10% are primary refractory, posing an unmet clinical need, especially in patients not eligible for hematopoietic stem cell transplant. Knowledge of DLBCL molecular pathogenesis has identified druggable molecular pathways. Surface antigens can be targeted by novel antibodies and innovative cell therapies.
Areas covered: This review illuminates those investigational drugs and cell therapies that are currently in early phase clinical trials for the treatment of DLBCL. New small molecules that modulate the pathways involved in the molecular pathogenesis of DLBCL, monospecific and bispecific monoclonal antibodies, drug-immunoconjugates, and cellular therapies are placed under the spotlight. A futuristic perspective concludes the paper.
Expert opinion: A precision medicine strategy based on robust molecular predictors of outcome is desirable in the development of investigational small molecules for DLBCL. Novel monoclonal and bispecific antibodies may be offered to (i) relapsed/refractory patients ineligible for CAR-T cells because of comorbidities, and (ii) younger patients before CAR-T cell infusion to reduce a high tumor burden. A focus on the optimal sequencing of the emerging DLBCL drugs is appropriate and necessary.
1. Introduction
Diffuse large B cell lymphoma (DLBCL) represents the most frequent type of B cell non-Hodgkin lymphoma (B-NHL) in the adult population worldwide [Citation1,Citation2]. DLBCL is characterized by a high degree of heterogeneity, both clinical and biological, and derives from mature B cells that have experienced the germinal center (GC) reaction and express potentially druggable B cell surface antigens, namely CD19, CD20, CD22, and CD79a/b [Citation1,Citation2]. According to the cell of origin (COO), DLBCL is distinguished into GC B cell-like (GCB-DLBCL), activated B cell-like (ABC-DLBCL), and unclassifiable [Citation1,Citation2]. This distinction has prognostic relevance with standard of care DLBCL treatment, and many studies have shown the superior outcome of GCB-DLBCL versus ABC-DLBCL when treated with the standard of care regimen, i.e. rituximab-CHOP (cyclophosphamide, doxorubicyne, vincristine, and prednisone) [Citation1–3].
Historically, the addition of rituximab to the CHOP regimen has represented a major step ahead in the treatment of DLBCL [Citation2]. However, despite the significant advances provided by the use of chemoimmunotherapy, 30 to 40% DLBCL eventually relapse and 10% are primary refractory [Citation2]. Relapsed and refractory (R/R) cases pose an unmet clinical need, especially in the case of patients who are not eligible for intensive therapeutic strategies based on hematopoietic stem cell transplantation. The clinical challenge posed by R/R DLBCL, the deeper progressive understanding of DLBCL biology and the knowledge that at least some of the molecular pathways involved in pathogenesis are druggable have prompted a large number of studies with new medicines, including small molecules, monoclonal antibodies (mAb) and cell therapies. The high degree of genetic complexity and the molecular heterogeneity of DLBCL represent a barrier toward a ‘one size fits all’ approach for novel treatments for DLBCL, as instead it has been the case in other types of B cell malignancies characterized by unifying genetic lesions or by the predominance of one specific molecular pathway. Conversely, the biological heterogeneity of DLBCL mandates the explorative targeting of multiple molecular pathways and prompts the search of biomarkers serving as predictors of response. This review will focus on investigational drugs and therapeutic approaches for DLBCL that are in early clinical development and have recently completed, or are currently undergoing, scrutiny in phase I or II studies. The literature search was performed on PubMed and main hematology meeting abstracts to obtain information on phase I and II clinical trials in DLBCL (last search date: 30 July 2020).
2. Molecular pathogenesis of DLBCL
The pathogenesis of DLBCL is a multistep process characterized by the accumulation of multiple genetic lesions, including gene mutations, chromosomal translocations, and other cytogenetic abnormalities [Citation3,Citation4]. Next-generation sequencing (NGS) studies in combination with other genetic approaches have unraveled the DLBCL genome in great detail, revealing on average 70 alterations in the exome per case, without a unifying genetic alteration occurring in all cases [Citation3,Citation4]. The mutational load of DLBCL is much higher when considering alterations of the whole genome, including noncoding regions, and not only the coding sequences. These notions may help understand the high degree of complexity of DLBCL biology and the marked heterogeneity of the disease presentation, response to treatment, and prognosis.
The alterations involved in DLBCL affect genes belonging to specific molecular pathways relevant for disease pathogenesis and implicated in epigenetic chromatin remodeling, immune escape, block of differentiation, cell proliferation, and constitutive activation of signal transduction pathways [Citation3,Citation4]. The study of the genomic landscape of DLBCL has also revealed differences in the mutational profile and in the molecular pathogenesis of GCB-DLBCL and ABC-DLBCL [Citation3,Citation4]. For example, genes involved in chromatin remodeling (EZH2, CREBBP, EP300, KMT2D) are more frequently mutated in GCB-DLBCL, whereas mutations of genes involved in NFKB activation and Toll-like receptor signaling (MYD88, CD79A/B, CARD11, BCL10, MALT1, TNFAIP3) are more frequently represented in ABC-DLBCL. Disruption of TP53, the hallmark of chemorefractoriness, occurs in both DLBCL categories.
Chromosomal translocations of BCL-6, BCL-2, and c-MYC are recurrent cytogenetic abnormalities in DLBCL [Citation1–4]. Chromosomal rearrangements of BCL-6, mapping to 3q27, occur in approximately 30% of DLBCL, and lead to deregulated expression of the bcl-6 protein that blocks GC B cell differentiation. Translocations of the anti-apoptotic gene BCL-2, mapping to 18q21, occur in 20–30% of DLBCL. Chromosomal translocations of band 8q24 occur in 10–15% DLBCL and target c-MYC, a common driver in human cancers regulating cell growth, proliferation, and survival. Both BCL-2 translocations and c-MYC translocations preferentially associate with GCB-DLBCL. The coexistence of BCL2 and c-MYC translocations confer a particular aggressiveness and a very severe prognosis to these cases (so called ‘double hit lymphomas’) which are recognized as an independent category and termed as high-grade B-cell lymphomas by the World Health Organization Classification of Lymphoid Neoplasms [Citation1].
3. Small molecule inhibitors
Recent understanding in the molecular pathogenesis of DLBCL has prompted the development of new small molecules targeting the pathways involved in lymphomagenesis with the aim to improve outcome and, in the future, refine a precision medicine approach to treatment ().
Figure 1. Molecular pathways in DLBCL and possible therapeutic targets for small molecule inhibitors. A plenty of molecular pathways and their genomic alterations have been described in DLBCL, contributing to initiation, maintenance and progression of the disease. Activation or deregulation of these molecular pathways can impair different cell mechanisms such as epigenetic control, proliferation, differentiation, and apoptosis. Even if some of these molecular pathways are actionable through different small molecules, the coexistence of numerous molecular alterations in the same tumor hinder the progress of precision medicine in DLBCL. Despite these limitations, some new small molecules are under development and seem promising in this field
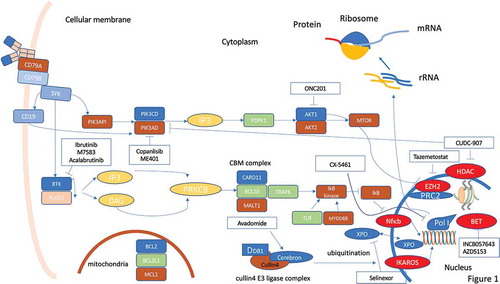
3.1. BTK inhibitors
The Bruton’s tyrosine kinase (BTK) is a component of the B cell receptor-mediated signaling machinery, and BTK inhibitors (BTKi) can effectively block B cell proliferation and survival (). BTKi have shown a significant activity in different categories of B cell malignancies, both indolent and aggressive and both treatment naïve and relapsed/refractory (R/R) [Citation5–12]. Initial results also suggested a certain activity of ibrutinib in R/R ABC-DLBCL [Citation13]. Subsequently, however, the phase III trial exploring R-CHOP with and without ibrutinib as first-line therapy in non-GCB-DLBCL has failed to show any significant advantage in the whole population [Citation14]. A sub-analysis of the trial has pointed to a potential advantage of the addition of ibrutinib in the younger population in terms of progression-free survival (PFS), event-free survival, and overall survival (OS). This result could be at least partially explained by the excess of toxicity reported in elderly patients [Citation14]. Despite the lower than expected efficacy of ibrutinib in DLBCL, a phase II trial in Richter syndrome with acalabrutinib, a second-generation BTKi, is ongoing [Citation15].
M7583 is a potent and highly selective BTKi, which is currently being investigated in a two-part, phase I/II trial in patients with R/R B cell malignancies, including DLBCL [Citation16]. The overall response rate (ORR) was 50% with a disease control of 78% and two patients achieving complete remission (CR) (). The most frequent toxicity was diarrhea in 33% of patients.
Table 1. Novel small molecules in DLBCL a
Overall, the addition of ibrutinib to the R-CHOP backbone awaits studies focusing on young DLBCL which are currently in progress. The availability of second and third-generation BTKi with a more favorable toxicity profile may improve the therapeutic index of BTKi addition to chemoimmunotherapy independent of age.
3.2. PI3K inhibitors
Phosphatidylinositol-3-kinase (PI3K) δ is responsible for the homeostasis and function of B cells and is involved in the interaction of the lymphoma clone with the tumor microenvironment. Although no data have been published to date in DLBCL treatment with the first-in-class PI3K inhibitor (PI3Ki), namely idelalisib, a few other PI3Ki are under investigation (). The PI3Kδi ME-401 has shown activity in indolent lymphoma but a lower efficacy in DLBCL with a 25% ORR in the rituximab combination group only [Citation17] (). A second PI3Ki under development in DLBCL is Copanlisib, a pan-class I PI3Ki with potent activity against PI3K-α and -δ isoforms. A first-in-human phase I study demonstrated a partial response (PR) in a patient with DLBCL (ORR 33%) [Citation18]. A phase II study in 48 patients with aggressive lymphoma, including 15 DLBCL unselected for COO, confirmed a 27% ORR rate [Citation19]. In a second phase II trial, 67 R/R DLBCL received copanlisib (ABC DLBCL, n = 19; GCB DLBCL, n = 30; unclassifiable, n = 3; missing, n = 15). The ORR was 19.4% (31.6% and 13.3% in ABC and GCB DLBCL, respectively) (). The ORR was similar in patients with/without CD79B mutations [Citation20]. Although the molecular pathophysiology of DLBCL suggests a significant impact of the PI3K pathway, results with PI3Ki in this lymphoma are less impressive when compared to indolent lymphoma. Of note, PI3Ki and especially copanlisib proved to be more effective in ABC-DLBCL and may perhaps have a future in the treatment in this disease subset.
3.3. BET inhibitors
Bromodomain and extraterminal (BET) proteins belong to a family of four epigenetic reader proteins (BRD2, BRD3, BRD4, and BRDT) recognizing acetyl groups in the histone tail and involved in recruiting transcriptional factors to activate gene transcription [Citation21] (). BET family proteins are involved in promoting aberrant oncogene expression in a variety of cancers. In particular, overexpression and gain-of-function mutations of BET proteins can alter gene transcription, histone modification, DNA repair, and apoptosis [Citation21].
INCB057643 is a selective small-molecule BET inhibitor. In a phase I study, a total of five lymphoma patients were enrolled, of which one achieved CR and two had stable disease [Citation22]. More recently, Falchook et al. reported the results of a phase I/II study with the BET inhibitors INCB054329 and INCB057643 [Citation23]. No responses were observed among the 13 R/R DLBCL (). AZD5153 is a potent, selective, and orally available BET/BRD4 bromodomain inhibitor possessing a bivalent binding mode [Citation24]. Unlike monovalent inhibitors, AZD5153 ligates two BRD4 bromodomains simultaneously. The enhanced avidity achieved through bivalent binding translates into increased antitumor activity in xenograft models of hematologic neoplasia, including DLBCL. The relationship between AZD5153 exposure and efficacy suggests that prolonged BRD4 target coverage is a primary efficacy driver. AZD5153 treatment markedly affects transcriptional programs of MYC, E2F, and mTOR [Citation24]. At the time of writing, the published data about BET inhibitors in DLBCL are too limited to draw firm conclusions. However, at least preclinical studies provide a rationale for the initiation of further trials in association with chemoimmunotherapy in the R/R setting.
3.4. Exportin inhibitors
Exportin 1 (XPO1) is one of eight nucleo-cytoplasmic shuttling proteins involved in protein export from the nucleus to the cytoplasm (). Its overexpression in DLBCL mediates the nuclear loss of tumor suppressor proteins in DLBCL and has been correlated with poor prognosis [Citation25,Citation26]. XPO1 blockade is able to revert the XPO1-mediated loss of multiple tumor suppressor proteins by forcing their nuclear retention [Citation27]. Selinexor is an oral selective inhibitor of XPO1-mediated nuclear export, already approved by FDA for advanced multiple myeloma in combination with dexamethasone, demonstrating activity in different lymphoproliferative diseases [Citation28,Citation29]. In particular, a phase I trial demonstrated a 32% ORR in R/R DLBCL with 10% CR [Citation28]. Recently, the results of the phase IIb clinical trial SADAL have been published [Citation30]. Two-hundred and sixty-seven patients were enrolled and randomized to two different doses of the drug (60 mg twice/week and 100 mg twice/week). The 100 mg arm was prematurely stopped, and all the analysis was carried on the population randomized to the 60 mg arm. The ORR reached 28% with 12% of patients achieving a CR [Citation30]. Based on these results, FDA granted accelerated approval for selinexor in adult R/R DLBCL which has failed at least two lines of therapy in June 2020.
3.5. Cereblon modulators
The cereblon modulator lenalidomide has high activity in R/R aggressive B-cell lymphomas [Citation31]. In untreated DLBCL, lenalidomide with R-CHOP21 has been documented to be safe and effective, particularly in non-GCB DLBCL [Citation32]. These results have led to the design of the ROBUST phase III trial comparing R-CHOP with and without lenalidomide in previously untreated ABC-DLBCL. However, in the ROBUST trial the primary endpoint was not met, although a trend for an improved PFS in patients with more advance disease (stage III/IV and IPI >3) was observed [Citation33]. Even though the ROBUST trial did not show any PFS advantage for ABC-DLBCL, a concurrent phase II trial of lenalidomide-R-CHOP in DLBCL suggested an improvement in PFS [Citation34]. This discrepancy may be at least partially explained by the different lenalidomide dose used in the two trials. On these grounds, novel cereblon modulators are also under scrutiny. In particular, avadomide (CC-122) is a cereblon-modulating agent exerting direct cell-autonomous activity against malignant B cells as well as immunomodulatory effects [Citation35] (). Upon binding to cereblon, avadomide promotes recruitment, ubiquitination, and subsequent proteasomal degradation of the hematopoietic transcription factors Ikaros (IKZF1) and Aiolos (IKZF3) [Citation36–38]. These molecular events lead to decreased proliferation and increased apoptosis of malignant B cells and costimulatory effects in T and natural killer cells (NK) [Citation39–41]. In preclinical models, avadomide has demonstrated antitumor activities in both ABC- and GCB-DLBCL [Citation35]. Among 84 de novo R/R DLBCL, the ORR was 29%, including 11% CR without differences in GCB- versus ABC-DLBCL () [Citation42]. On the other hand, two different subgroups with impact on efficacy endpoints were identified based on GEP: classifier positive, characterized by an enriched infiltration of T cells and macrophages; and classifier negative, associated with a predominance of intratumoral B cells. Consistent with the proposed mechanism of action of avadomide on the microenvironment, classifier-positive DLBCL patients showed improved ORR and PFS compared to cases with a predominance of intratumoral B cells [Citation42].
Although only limited data are available, cereblon modulators seem to be promising in DLBCL. However, avadomide showed to be efficacious only in a subgroup of patients, who cannot be identified by standard molecular and histochemical classification. This may hamper the identification of the best candidates for the treatment in the clinical practice, leading to an improper use of the drug, unless a specific GEP-based classification is implemented.
3.6. EZH2 inhibitors
Epigenetic modulation of histones plays a critical role in oncogenic transformation in many malignancies and is an area of intense clinical research. The genes encoding chromatin-modifying proteins are frequently targeted by DNA mutations in B-cell NHL deriving from GC cells and including DLBCL [Citation43,Citation44]. The methyltransferase encoded by the EZH2 gene makes part of the PRC2 (for Polycomb Repressive Complex 2) complex and can methylate histone 3 lysine 27 (H3K27), generating the histone mark H3K27me3 that favors repression of transcription (). In lymphoid development, EZH2 downregulates the expression of genes regulating cell cycle and differentiation and is counteracted by the SWI/SNF (Switch/Sucrose NonFermentable) multiprotein complex that also participates in chromatine remodeling [Citation45,Citation46]. Gain of function mutations of EZH2 are reported in 20% of GC B cell lymphoma, resulting in an aberrant proliferative dependency on EZH2 activity and disruption of the differentiation process through hyper-trimethylation of H3K27 [Citation43–46].
Recently, a phase Ib trial combining R-CHOP with the EZH2 inhibitor tazemetostat, which already gained FDA approval for EZH2 mutated R/R follicular lymphoma in June 2020, has been performed in newly diagnosed DLBCL () [Citation47]. Grade ≥3 toxicities were constipation, nausea, hypokalemia, and hematologic cytopenias. No mature data on PS and PFS were reported due to the very short follow-up of the study. The same agent has been used also in the R/R setting in a phase I trial on 64 patients including 13 DLBCL [Citation48]. Grade 4 thrombocytopenia was the single dose-limiting toxicity, with no treatment-related deaths. Durable objective responses were observed in 4 DLBCL, namely 1 CR and 3 PR [Citation48].
A future step in inhibiting this pathway will be the use of dual EZH1 and EZH2 inhibitors. Recently, a novel dual EZH1/EZH2 inhibitor has been tested in vivo and in vitro and has shown greater antitumor efficacy than EZH2 selective inhibition against DLBCL cells harboring gain-of-function EZH2 mutations [Citation49]. Overall, tazemetostat and dual inhibitors may represent a new avenue for combination therapy of DLBCL.
3.7. Akt/ERK inhibitor
ONC201 is the founding member of the imipridone class of small molecules, and induces caspase-dependent apoptosis also via inhibition of Akt phosphorylation [Citation50] (). ONC201 is currently being evaluated in advanced cancer patients, including DLBCL (). A phase I/II trial in different subtypes of NHL is still recruiting, and no data have been published at the time of writing.
3.8. Dual HDAC and PI3K inhibitor
CUDC-907, a dual-acting inhibitor of both class I and II histone deacetylase (HDACs) and class I PI3Ks, was shown to synergistically downregulate MYC protein levels and to induce apoptosis in ‘double-hit’ DLBCL cells () [Citation51]. CUDC-907, effectively suppresses the growth and survival of MYC-altered or MYC-dependent cancer cells, such as double hit DLBCL, and MYC protein downregulation is an early event induced by CUDC-907 treatment. In a phase I trial, CUDC-907 was orally administered to heavily pre-treated adult patients with lymphoma or multiple myeloma [Citation52]. Five out of 9 DLBCL patients achieved objective responses (2 CR, 3 PR) occurring also in transformed DLBCL. These data prompted a second phase I trial in R/R DLBCL, with a particular focus on those with MYC-altered disease, with or without Rituximab [Citation53]. Thirty-seven patients (25 in mono-therapy arm, 17 in combination arm) were enrolled, among which 14 had confirmed MYC-altered disease. The ORR was 37% (). The ORR in MYC-altered DLBCL patients was 64% (7 out of 11; 4 CR and 3 PR), while it was 29% in MYC unaltered DLBCL, and 17% in those with unknown MYC status. Median duration of response (DOR) was 13.6 months in MYC-altered patients compared to 6.0 months in MYC-unaltered DLBCL. In the challenging setting of relapsed MYC translocated DLBCL, CUDC-907 achieved a higher than expected ORR with a fairly good DOR [Citation53]. These data should lead to plan a further development of the drug.
3.9. RNA polymerase transcription inhibitors
Ribosomes are fundamental for growth and proliferation both in normal cells and in cancerous cells. Their biogenesis is strictly related to the transcription of ribosomal RNA and so ultimately to the activity of RNA Polymerase I (Pol I). The activity of Pol I is regulated by different pathways such as RAS, MYC, and PI3K, which can be deregulated in DLBCL, making the inhibition of Pol I a logical therapeutic strategy [Citation54–57]. CX-5461 is a small-molecule inhibitor of Pol I transcription, which, inhibiting rDNA transcription, elicits apoptosis through the nucleolar stress pathway in a p53 dependent or independent manner [Citation58]. In a phase I study, 16 patients, including four R/R DLBCL, received CX-5461 [Citation58]. The best ORR was stable disease in 2 out of 4 DLBCL. The drug was well tolerated with exclusively cutaneous AEs.
4. Monoclonal antibodies (mAbs)
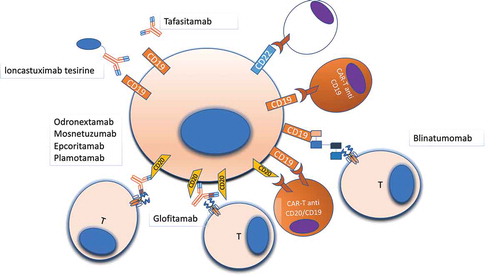
After more than 20 years of clinical experience, it is worth evident that the introduction of anti-CD20 treatment has represented a major milestone in the treatment of DLBCL [Citation59–62]. Despite these significant improvements, 30% to 40% DLBCL eventually relapse and 10% are primary refractory. Outcome in those patients is poor, resulting in an ORR of 27% to 63% with long survival of 48% [Citation63–65]. In this clinical setting, targeting new surface antigens or using drug immunoconjugates, as well as harassing the immune system with bispecific antibodies, are appealing treatment approaches ().
Figure 2. Druggable surface molecules in DLBCL. Surface antigens are the most reachable part of the cell, and monoclonal antibodies and cellular therapies targeting surface antigens represent an important therapeutic strategy in lymphomas. Several different antibodies, with different mechanisms of action, and CAR-T cells are under development for treating DLBCL, with the majority targeting CD19, CD20, CD22 due to the relative abundance of all these antigens on the cell surface. Other antigens such as CD79b (not shown) have been already used as target of ADCs (Polatuzumab-vedotin)
4.1. Monospecific antibodies
Monospecific mAbs target one specific cell surface antigen and exert their cytotoxic activity either directly or through immune-mediated mechanisms.
4.1.1. Tafasitamab
Tafasitamab (MOR208) is an Fc-engineered anti-CD19 humanized antibody (). In a phase II trial of R/R NHL, Tafasitamab was able to attain an ORR and CR rates of 26% and 6%, respectively, with a DOR of 20.1 months [Citation66]. After a median follow-up of 21 months, the median PFS was 2.7 months. These unsatisfactory results led to a second phase II trial in which Tafasitamab was associated with lenalidomide [Citation67]. The trial enrolled 81 relapsed DLBCL patients not eligible for autologous transplant. The ORR was 58% with 33% CR. At 12 months follow-up, PFS was 16.3 months while OS and DOR were both not reached [Citation67]. On these grounds, tafasitamab in combination with lenalidomide has recently been approved by FDA in R/R follicular lymphoma and R/R DLBCL not eligible for autologous stem cell transplant.
4.2. Antibody-drug conjugates
Antibody-drug conjugates (ADCs) are mAbs capable of targeting cell surface antigens with high specificity and delivering therapeutic agents to tumor cells. The cytotoxic drug is conjugated to the mAb through a stable linker ensuring that the drug does not detach from the antibody.
4.2.1. Polatuzumab vedotin
Polatuzumab is an approved drug immunoconjugate targeting CD79b. The antibody is conjugated through a protease cleavable linker to monomethyl auristatin E (MMAE). Upon binding to the target, polatuzumab is internalized and is cleaved by lysosomal proteases to release MMAE. This agent binds to microtubules interfering with cell division and leading to apoptosis. The drug has already been granted FDA and EMA approval for R/R DLBCL not eligible for autologous stem cell transplant in combination with anti CD20 (rituximab or obinutuzumab) and bendamustine based on the results of a phase Ib/II trial [Citation68,Citation69].
4.2.2. Loncastuximab tesirine
Loncastuximab tesirine (ADCT-402) is a novel CD19-targeted drug immunoconjugate delivering tesirine (SG3199), a highly cytotoxic DNA cross-linking pyrrolobenzodiazepine dimer (PBD) warhead showing potent and highly targeted in vitro cytotoxicity in CD19-expressing human cell lines () [Citation70]. In a recent phase I trial, 85 R/R NHL were treated with loncastuximab tesirine at a dose 15 to 200 mcg/kg [Citation71]. The ORR at doses ≥120 μg/kg was 59.4% (40.6% CR; 18.8% PR). Median DOR, PFS, and OS (all doses) were 4.8, 5.5, and 11.6 months, respectively [Citation71].
4.2.3. Coltuximab ravtansine (SAR3419)
Coltuximab ravtansine is an anti-CD19 ADC loaded with a potent cytotoxic may-tansinoid, DM4, via an optimized, hindered, disulfide bond. The drug has been tested as monotherapy in a phase I trial, enrolling 41 R/R DLBCL, showing an encouraging 44% ORR with a good safety profile [Citation72]. However, in a phase II trial, the benefit for the treatment was confirmed only in the relapsed subpopulation, but not in the refractory cohort [Citation73].
4.2.4. Pinatuzumab vedotin (DCDT2980S)
Pinatuzumab vedotin is an ADC targeting CD22 loaded with MMAE. Clinical activity and an acceptable safety profile have been already reported in R/R NHL in a phase I trial as monotherapy and in association with rituximab [Citation74]. Similar results were recently reported in a randomized, multicenter, open-label study, comparing pinatuzumab monotherapy with pinatuzumab + rituximab [Citation75]. Median PFS reached 115 days for DLBCL patients both in monotherapy and in association with rituximab with a 40% ORR in both arms [Citation75]. A further trial compared rituximab-pinatuzumab vs rituximab-polatuzumab [Citation76]. Limiting the analysis to the R/R DLBCL cohort, the rituximab-pinatuzumab arm proved to be at least non-inferior to the rituximab-polatuzumab arm with an ORR of 60% with 26% CR [Citation76].
4.3. Bispecific antibodies
Exploiting cytotoxic T-cells as a therapeutic tool against tumor cells has been considered as a therapeutic strategy for many cancers, including DLBCL [Citation77]. Utilizing bispecific Abs (bsAbs), the autologous patient T cells can be redirected against DLBCL and, in this manner, several anti-tumor cytotoxic mechanisms can be activated [Citation78]. The definition of bsAbs comprises a large group of molecules engineered to recognize two different epitopes or antigens on the target cell [Citation78].
4.3.1. Blinatumomab
Blinatumomab is a T cell-engaging bsAb simultaneously targeting the CD3 T cell antigen and the pan-B CD19 antigen () [Citation79]. The dual binding is constructed from a CD19 specific single-chain antibody and an anti-CD3 portion [Citation79]. The cell lysis by blinatumomab occurs via multiple mechanisms, including direct binding to CD19 and secretion of granzymes and perforins from the synapse between T cells and target B cells [Citation80]. Seminal evidence for the use of blinatumomab in DLBCL is based on a phase I trial, including 76 heavily pre-treated NHL patients [Citation81]. A dose-dependent response was evident with no responses with blinatumomab dosing <15 μg/m2/day. At the 60 μg/m2/day dose, the ORR was 69% and CR/complete remission unconfirmed (CRu) was 37% with long-term remissions independent of prior therapies and histologic subtype [Citation81]. The most common grade ≥3 adverse events (AE) were lymphopenia (69%) and grade neurologic events (22%). All neurological toxicities, mainly related to cytokine release syndrome (CRS), developed after a median of 2 days of treatment and resolved after drug discontinuation [Citation81]. These initial results prompted a phase II trial in R/R DLBCL [Citation82]. The trial compared a stepwise or flat dosing of blinatumomab by continuous infusion. Among 21 evaluable patients, the ORR after 1 blinatumomab cycle was 43%, with a CR in 19%. As expected, grade ≥3 neurologic events were observed in a sizable fraction of patients. Most neurologic events finally resolved in the stepwise dosing cohort. Conversely, the flat-dose cohort was stopped because of grade ≥3 neurologic events [Citation81]
4.3.2. Glofitamab (CD20 TCB or RG6026)
The relevance of CD20 targeting in DLBCL was known since the early phases of rituximab. Starting from the ‘proof-of-principle’ concept of bsAb exemplified by the anti-CD19 blinatumomab, the CD20 antigen has been exploited for designing novel bsAb (). Glofitamab is a new bsAb with IgG-like pharmacokinetic properties and a unique ‘2:1’ structure [Citation83,Citation84]. The molecule comprises two CD20 binding Fabs (derived from the Type II CD20 IgG1 obinutuzumab), one CD3ε binding Fab (fused to one of the CD20 Fabs via a short flexible linker), and an engineered, heterodimeric Fc region with completely abolished binding to FcγRs and C1q () [Citation83]. In a phase I, first in human trial, 47 R/R aggressive and 17 R/R indolent lymphomas received glofitamab at doses ranging from 5 μg to 1800 μg every 2 weeks. CR was noted from 300 μg dose onwards after two cycles of therapy in 29 evaluable patients. ORR was 38% with 24% CR rate, and all CRs were sustained at a median follow-up of 96 days () [Citation84]. The results of an expansion phase I/II trial and two different combination trial (with the anti-CD20 obinutuzumab and the anti-PD-L1 atezolizumab) are reported in [Citation85–87]. The toxicity profile is reported in . All CRS events were manageable with no central nervous system toxicity.
Table 2. Efficacy endpoints for different bispecific antibodies and CAR-T cells a
Table 3. Toxicity profile of bispecific antibodies and CAR-T cell treatments for DLBCLa
4.3.3. Odronextamab
Odronextamab (REGN1979) is a fully humanized bispecific IgG4 Ab targeting CD20 and CD3 and designed to resemble natural human Abs () [Citation88]. This bsAb induces prolonged B-cell depletion in the peripheral blood as well as in lymphoid organs in preclinical models [Citation88]. In a phase I study on R/R NHL, 25 heavily pretreated patients including 12 DLBCL were treated with a flat dose [Citation89]. In this small trial, the ORR of DLBCL was less impressive (improvement of the target lesion observed in 5 out of 12 pts) compared to follicular lymphoma (FL) that had the best ORR (almost 100%) (). The toxicity profile of the drug is reported in . This phase I study led to a phase II study limited to R/R FL.
4.3.4. Mosunetuzumab
Mosunetuzumab is a humanized, bispecific antibody binding to CD20 on lymphoma cells and to CD-3ε expressed by T cells (). The first clinical results with mosunetuzumab were recently reported: in R/R aggressive NHL (141 cases, of which 87 de novo DLBCL and 29 transformed DLBCL), the ORR was 37.1%, with a CR rate of 19.4% () [Citation90]. Grade ≥3 neurotoxicity and CRS were reported in 5% of cases ().
4.3.5. Epcoritamab
Epcoritamab (GEN3013) is the first subcutaneous bsAb that targets CD3 and CD20 [Citation91]. Eighteen heavily pre-treated patients (14 DLBCL) were enrolled into the dose-escalation part of the phase I trial. Efficacy data are represented in . Despite increasing epcoritamab dosage, all CRS events were graded as non-severe (G1-G2). These encouraging safety data led to a phase II trial.
4.3.6. Plamotamab
Plamotamab (XmAb13676) is a humanized bsAb binding CD20 and CD3. The results of the first-in-human trial have been recently presented [Citation92]. Among 44 subjects who have been treated, there have been 7 objective responses, including 2 CR and 3 PR in DLBCL. The most common toxicity was CRS, which occurred in 11.1% subjects with 1 of the events being Grade 4 and the other events being ≤ Grade 2 [Citation92].
5. Cellular therapies
Cellular therapies for DLBCL range from CAR-T cells, either monospecific or bispecific, to the combination of CAR-T cells with agents targeting the microenvironment, and to engineered T-cells carrying an antibody T cell receptor (TCR). Whereas monospecific CAR-T cells are already available for clinical practice, other cellular therapies are still in clinical development.
5.1. CAR-T cells targeting one single antigen
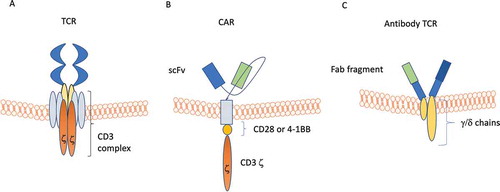
Tumor regression can be mediated by transfer of major histocompatibility complex (MHC)–restricted T cells recognizing tumor-associated antigens. This notion has prompted the development of strategies of adoptive cell therapy (ACT). These attempts have faced the issue of obtaining adequate numbers of T cells with defined specificity and MHC restriction for individual patients. A major step ahead to overcome these difficulties has been represented by the technology allowing the transfer of chimeric antigen receptors (CARs) into T-cells [Citation93]. Briefly, a CAR is a transmembrane protein build up by: i) a single-chain immunoglobulin-derived variable fragment (scFv) targeting the chosen antigen; ii) a transmembrane hinge; iii) an intracellular signaling domain consisting of CD3ξ and co-stimulatory molecules such as CD-28 and 4–1BB (). Such a receptor offers major advantages, since: i) it is fully independent from HLA, circumventing cancer-induced HLA reduced expression; ii) costimulatory molecules in the transduction tail improve T-cell proliferation, cytokine production and long-term persistence. Approved CAR-T cell therapies target CD-19 and include (): tisagenlecleucel for recurrent pediatric acute lymphoblastic leukemia (ALL) and R/R large B cell lymphoma [Citation94]; axicabtagene ciloleucel for R/R large B cell lymphoma [Citation95,Citation96]; lisocabtagene maraleucel for R/R DLBCL [Citation97,Citation98]. The efficacy data of CAR-T cells in DLBCL are reported in .
Figure 3. Schematic views of normal T cell receptor (TCR), chimeric antigen receptor (CAR), and antibody TCR. Panel A: TCR is composed of a binding outer domain that recognizes the antigen presented by antigen-presenting cells through MHC molecules. This interaction leads to the activation of different cellular pathways only in the presence of a costimulatory signaling through CD28/C4/C8/CD45. Panel B: CAR is composed of a scFV targeting antigen on cancer cells, a transmembrane linker and an inner portion containing both CDξδand the costimulatory CD28/4-1BB domain. This type of receptor is able to induce T-cell activation in an MHC-independent manner. Panel C: antibody TCR contains an intact γ/δ chain as transmembrane and intracellular domains, while the recognition domains is a Fab fragment targeting one specific antigen. Similar to CAR, also antibody TCR acts in an MHC-independent manner but retains the autoregulatory inhibition pathways similar to the normal TCR
As outlined in , the major safety concerns of CAR-T cells are related to life-threatening CRS and neurologic dysfunction, which are mainly related to the rapid expansion of T cells and to the consequent cytokine storm [Citation94,Citation95,Citation97]. Overall, grade ≥3 CRS (13–14%) and neurotoxicity (7–28%) have been reported in all CAR-T cell studies for R/R DLBCL. Different severity scoring systems were used in clinical trials making a comparison challenging.
5.2. Bispecific CAR-T
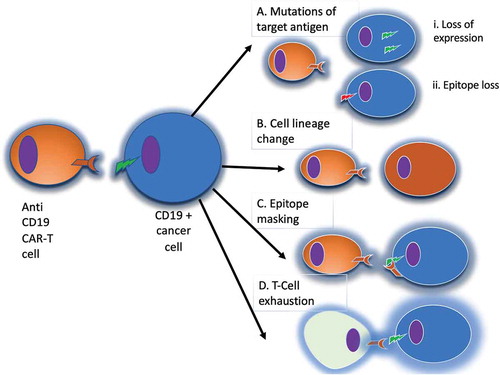
A fraction of lymphoma patients fails to respond to CAR-T cells, and other relapse after an initial response. Among the known escape mechanisms to CAR-T cells (), the best identified mechanism of disease relapse is target antigen loss, namely CD19 [Citation99,Citation100]. One obvious way to overcome the problem of antigen loss following CAR-T cell therapy is targeting more than one antigen (). Pre-clinical data in support of using dual-targeting in B cell malignancies have emerged, demonstrating the effectiveness of such approach in different mouse models [Citation101–103].
Figure 4. Mechanisms of evasion from anti-CD19 CAR-T cells. Panel A: in the case of loss of the target antigen, two different mechanisms have been described: i) internalization of the surface antigen (e.g. CD19); ii) amino-acidic modification of the CAR binding site. Panel B: in the case of cell lineage change, tumor cells can switch their phenotype to a different lineage, that constitutively does not express CD19. Panel C: epitope masking occurs when during lentiviral transfection a single leukemic or lymphoma cell is modified to express the CAR. Panel D: T-cell exhaustion may occur when the CAR-T cell population is chronically stimulated by the targeted antigen, leading to reduced T cell cytotoxic activity
Recently, a phase I bispecific CAR-T cell trial with an anti-CD19/anti-CD20 tandem receptor has demonstrated achievement of CR or PR in 3/6 heavily pre-treated and relapsed aggressive B cell NHL [Citation104]. Interestingly, among the three patients who progressed or relapsed, all retained either CD19 or CD20 positivity on the subsequent biopsy, suggesting other escape mechanisms rather than antigen loss [Citation104]. Similar to the development of a CD20-CD19 CAR-T cell, a bispecific CD19-CD22 CAR-T cell has been developed [Citation105]. In the phase I study, this bispecific CAR-T cell has been evaluated in seven patients of which five had DLBCL [Citation106]. Among patients with DLBCL, the ORR was 80% with a 40% CR rate (). No grade ≥3 AE were reported, although six patients developed reversible CRS and neurotoxicity [Citation106].
Table 4. New cellular technologies under development for relapsed/refractory DLBCL a.
5.3. Strategies to overcome the microenvironment
Whichever the target, CAR-T cells achieve an ORR in lymphomas which is lower than in ALL. This lower ORR might be related to the presence of an immunosuppressive microenvironment, hampering CAR-T function. In order to overcome this possible immunomodulation, a phase I trial was designed based on the association of bicistronic CD19-CD22 CAR-T followed by consolidation with pembrolizumab [Citation107]. Six R/R DLBCL and two transformed DLBCL were treated with this novel approach. Four out of five evaluable patients responded with an ORR of 80% and a CR rate of 40%. Escalation to higher doses and updated follow-up of patients is planned.
Another possible way to overcome the microenvironment hampering effect is to enhance the proliferative potential and the interleukin production. The anti-CD-19 (19–28z/4-1BBL) ‘armored’ CAR-T cells have been engineered with both CD-28 and 4–1BB co-stimulation for increasing tumor removal, continued T cell proliferation, and persistence [Citation108]. In a phase I trial, 25 patients with R/R NHL, including de novo and transformed DLBCL, received varying doses of ‘armored’ CAR-T cells [Citation109]. Fifty-seven % of patients achieved CR, and at a median follow-up of 93 days, almost all patients remained in CR. Sixty-seven % of patients experienced grade 1–2 CRS without any severe CRS.
5.4. Engineered T-cells
Both CRS and neurological dysfunction are related to the expansion and activation of CAR-T cells and the consequent cytokine storm [Citation110,Citation111]. One possible approach to reduce severe cytokine-mediated toxicities is to channel activation signaling through an endogenous CD3 complex by replacing the antigen recognition domain of a γδTCR with an antibody-derived Fab fragment. In this way, it may be possible to create a synthetic receptor that uses endogenous TCR signaling pathways with the ability to directly target an extracellular antigen in an HLA-independent manner [Citation111]. This platform was named ARTEMIS and it has been coupled with a human anti-CD-19 antibody (ET190L1) to produce an engineered autologous T cell [Citation111,Citation112]. In vitro, the re-engineered complex retains the potency against different lymphoproliferative disease cell lines and has shown a significant reduction in cytokine release during antigen-specific T cell activation [Citation112]. In the first-in-human trial, 21 R/R NHL were treated with one ET190L1-ARTEMIS T cell infusion at different doses. The ORR was 52% at one month, with 55% complete CR and 45% PR (). At 6 months, 5/6 CR were still in CR [Citation113,Citation114]. Of interest, no severe AEs leading to treatment discontinuation, CRS, or neurotoxicity were reported [Citation114].
5.5. NK cell therapies
The interest in NK cells arises from the evidence of their significant involvement in graft versus leukemia/lymphoma without increasing or even preventing graft versus host disease [Citation115–117]. On the contrary, autologous NK lymphocytes proved to be of limited value, even after ‘ex vivo’ expansion and activation, in relapsed multiple myeloma, demonstrating the importance of a mispairing between KIRs and their ligand [Citation118]. The further step was to assess the possibility to use cord-derived NK cells. A phase I trial demonstrated the safety of such an approach in multiple myeloma [Citation119], opening the way to adoptive immune therapy exploiting the engineering of cord-derived NK cells with CARs. This step may allow to produce a readily available product, without the need to create a specific CAR for every patient. In a phase I trial of R/R lymphoproliferative diseases (three transformed follicular lymphomas, 2 DLBCL, 1 follicular lymphoma grade 3B, 3 CLL, and 2 Richter transformation), anti-CD19-CAR-NK cells derived from cord blood showed a 73% ORR [Citation120]. Interestingly, no significant toxicities were reported, and in particular no CIRS, neurotoxicity, or graft-versus-host disease. Even though preliminary, these results appear promising and warrant further evaluation especially considering the possibility to obtain an off-the-shelf product.
6. Conclusions
The deeper understanding of DLBCL molecular pathogenesis has revealed several molecular pathways druggable with small molecules that have completed or are currently undergoing scrutiny in phase I or II clinical trials for R/R patients. If successful and if safety allows, these drugs may also be combined to standard of care chemoimmunotherapy for first-line treatment of DLBCL. In parallel, new monoclonal antibodies have emerged, either directed against antigens (CD19) not previously targeted in DLBCL, or acting as ADCs to deliver potent drugs to lymphoma cells, or recruiting cytotoxic T cells against the DLBCL clone thanks to their bispecific configuration. The advent of CAR-T cell therapy has opened a new avenue for DLBCL therapy, and novel approaches to activate the autologous immune system against DLBCL are being continuously proposed and tested. Despite promising signals in early phase trials dedicated to R/R patients, the experience gained until now in phase III trials comparing R-CHOP versus (R-)CHOP with new antibodies or small molecules inhibiting a specific pathway suggest that it may not be easy to achieve a superiority versus the current standard of care in the context of first-line treatment of DLBCL.
7. Expert opinion
The concept of precision medicine applied to neoplastic disorders implies the individual tailoring of management and treatment of the disease based on the tumor genomic landscape, coupled with clinical features and comorbidities of the host. Importantly, pursuing the strategy of precision medicine has been highly successful in some B cell malignancies, as exemplified by the case of CLL [Citation121,Citation122]. In this respect, the high degree of molecular complexity of DLBCL may represent a serious challenge toward a ‘one size fits all’ treatment approach, especially in the case of drugs targeting one specific pathway, and may require a precision medicine strategy based on molecular biomarkers serving as robust predictors of response and long-term outcome [Citation2–4]. Although precision medicine is one major focus of current research in DLBCL, the results achieved until now are not yet mature for translation into the clinical practice and will require future efforts. One important requirement is represented by the availability of robust and clinically meaningful biomarkers that may predict treatment outcome and may help clinicians in choosing the best therapeutic option. These molecular predictors should be easily and reliably assessable in the everyday clinical practice and should be validated across different centers.
Due to clonal evolution, the genotypic landscape of DLBCL may differ at different anatomic sites, further increasing the genomic complexity of the disease and posing an additional challenge to a precision medicine strategy, that might not be fully informative if based on one single site of the disease. Liquid biopsy analyzing the circulating tumor DNA and capturing genetic features also at distance from the initial biopsy site may represent a useful tool for gaining a comprehensive view of the DLBCL genome in an individual patient and for better targeting the treatment choices based on biomarkers [Citation123,Citation124]. In addition, liquid biopsy may provide a valid approach for early assessment of response to a given drug and for monitoring minimal residual disease upon a given treatment [Citation125,Citation126]. The integration of liquid biopsy in the study of DLBCL investigational drugs and cell therapies may pave the way to new concepts in disease targeting and monitoring.
Whereas target therapy with small molecules is not yet fully mature for clinical practice in DLBCL, new mAbs, and in particular bsAbs, appear to be promising in R/R DLBCL cases in terms of efficacy and safety (). On these grounds, bsAbs may represent a good alternative to the use of CAR-T cells, at least in subsets of patients, although direct comparative data are obviously lacking (). Bearing these concepts in mind, and in the absence of data regarding the impact on OS, we may envisage two possible modalities for the use and sequencing of novel mAbs and of bsAbs. A first scenario entails the adoption of bsAbs in DLBCL not eligible for CAR-T cells due to patient comorbidities, which have recently been reported as the major limitation in a real life CAR-T cell study [Citation127]. A second scenario is represented by young R/R DLBCL patients with high tumor burden, who are candidates for CAR-T cell therapy. In fact, a high tumor burden before the infusion of CAR-T cells is a predictor of a higher rate of cytokine related AEs and of lower efficacy of cellular therapy [Citation127]. Using these novel antibodies, especially bsAbs targeting CD20, as a preparatory regimen might result in reduction of the toxicities and improved efficacy of CAR-T cells, mainly related to a better tumor control.
The number of CAR-T cell products is continuously expanding, and all the single cells have relevant manufacturing and functional differences. Notably, the main reason for developing new cellular products is to reduce the incidence of severe neurotoxicity and CRS, without reducing the response rate. Armored CAR-T and CD19/CD22 targeting CAR-T cells seem to be promising cellular products. With an ORR exceeding 50–60% and a more favorable toxicity profile across the different phase I/II trials, these new cellular technologies are at least comparable with the available anti-CD19 CAR-T cells (). Moreover, the use of multiple targeted agents concurrently has the potential to reduce the recurrence rate after CAR-T cell therapy and, consequently, to increase the long-term effect of the treatment. These clinical advances, if corroborated by future studies, may potentially widen the spectrum of DLBCL patients who might benefit from cellular therapies.
Finally, in face of the many investigational drugs under clinical trial, it should be always kept in mind that the development of new treatments for DLBCL, as for most if not all cancers, should consider sustainability and equity in the access to novel life-saving drugs for all those who are in medical need.
Article Highlights
Thirty to 40% of DLBCL patients relapse [with 10% being primary refractory]; this poses an unmet clinical need especially in patients who are ineligible for intensive therapeutic strategies.
Recent understanding in the molecular pathogenesis of DLBCL has prompted the development of new small molecules that target the pathways involved in lymphomagenesis.
Targeting new surface antigens or using drug immunoconjugates, and harnessing the immune system with bispecific antibodies, are emerging treatment approaches.
Monospecific CAR-T cells are already available in clinical practice, and several other cellular therapies are in clinical development.
A true precision medicine approach to DLBCL requires the identification of robust predictors that will allow a rational choice among the therapeutic options under development in the DLBCL therapeutic landscape.
This box summarizes key points contained in the article.
Declaration of interest
A Patriarca discloses roles in advisory boards of Ariad, Sanofi and Takeda; G.G. discloses roles in advisory boards or speaker bureaus of Abbvie, AstraZeneca, Janssen and Sunesys. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016 May 19;127(20):2375–2390.
- Li S, Young KH, Medeiros LJ. Diffuse large B-cell lymphoma. Pathology. 2018 Jan;50(1):74–87.
- Pasqualucci L, Dalla-Favera R. Genetics of diffuse large B-cell lymphoma. Blood. 2018 May 24;131(21):2307–2319.
- Miao Y, Medeiros LJ, Li Y, et al. Genetic alterations and their clinical implications in DLBCL. Nat Rev Clin Oncol. 2019 Oct;16(10):634–652.
- Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013 Aug 8;369(6):507–516.
- Byrd JC, Brown JR, O’Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014 Jul 17;371(3):213–223.
- Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015 Dec 17;373(25):2425–2437.
- Byrd JC, Harrington B, O’Brien S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016 Jan 28;374(4):323–332.
- Noy A, de Vos S, Thieblemont C, et al. Targeting bruton tyrosine kinase with ibrutinib in relapsed/refractory marginal zone lymphoma. Blood. 2017 Apr 20;129(16):2224–2232.
- Dimopoulos MA, Tedeschi A, Trotman J, et al. Phase 3 trial of ibrutinib plus rituximab in waldenström’s macroglobulinemia. N Engl J Med. 2018 Jun 21;378(25):2399–2410.
- Wang M, Rule S, Zinzani PL, et al. Durable response with single-agent acalabrutinib in patients with relapsed or refractory mantle cell lymphoma. Leukemia. 2019 Nov;33(11):2762–2766.
- Owen RG, McCarthy H, Rule S, et al. Acalabrutinib monotherapy in patients with waldenstrom macroglobulinemia: a single-arm, multicentre, phase 2 study. Lancet Haematol. 2020 Feb;7(2):e112–e121.
- Wilson WH, Young RM, Schmitz R, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015 Aug;21(8):922–926.
- Younes A, Sehn LH, Johnson P, et al. Randomized phase III trial of ibrutinib and rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non-germinal center B-cell diffuse large B-cell lymphoma. J Clin Oncol. 2019 May 20;37(15):1285–1295.
- Appleby N, Eyre TA, Cabes M, et al. The STELLAR trial protocol: a prospective multicentre trial for richter’s syndrome consisting of a randomised trial investigation CHOP-R with or without acalabrutinib for newly diagnosed RS and a single-arm platform study for evaluation of novel agents in relapsed disease. BMC Cancer. 2019 May 20;19(1):471.
- Jurczak W, Rule S, Townsend W, et al. A phase I/II, first in human trial of the bruton’s tyrosine kinase inhibitor M7583 in patients with B-cell malignancies. Blood. 2018;132(Supplement 1):4161.
- Pagel John RN, Jagadeesh D, Stathis A, et al. The PI3KΔ inhibitor ME-401 is well-tolerated on intermittent schedule and produces a high-rate of durable responses in relapsed/refractory (R/R) indolent B-cell malignancies. Hemisphere. 2020;4(S1):546.
- Patnaik A, Appleman LJ, Tolcher AW, et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann Oncol. 2016 Oct;27(10):1928–1940.
- Dreyling M, Morschhauser F, Bouabdallah K, et al. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann Oncol. 2017 Sep 1;28(9):2169–2178.
- Lenz G, Hawkes E, Verhoef G, et al. Single-agent activity of phosphatidylinositol 3-kinase inhibition with copanlisib in patients with molecularly defined relapsed or refractory diffuse large B-cell lymphoma. Leukemia. 2020 Aug;34(8):2184-2197. .
- Reyes-Garau D, Ribeiro ML, Roué G. Pharmacological targeting of BET bromodomain proteins in acute myeloid leukemia and malignant lymphomas: from molecular characterization to clinical applications. Cancers (Basel). 2019 Oct 2;11(10):1483.
- Forero-Torres A, Rosen S, Smith DC, et al. Preliminary results from an ongoing phase 1/2 study of INCB057643, a bromodomain and extraterminal (BET) protein inhibitor, in patients (pts) with advanced malignancies. Blood. 2017;130(Supplement 1):4048.
- Falchook G, Rosen S, LoRusso P, et al. Development of 2 bromodomain and extraterminal inhibitors with distinct pharmacokinetic and pharmacodynamic profiles for the treatment of advanced malignancies. Clin Cancer Res. 2020 Mar 15;26(6):1247–1257.
- Rhyasen GW, Hattersley MM, Yao Y, et al. AZD5153: A novel bivalent BET bromodomain inhibitor highly active against hematologic malignancies. Mol Cancer Ther. 2016 Nov;15(11):2563–2574.
- Culjkovic-Kraljacic B, Fernando TM, Marullo R, et al. Combinatorial targeting of nuclear export and translation of RNA inhibits aggressive B-cell lymphomas. Blood. 2016 Feb 18;127(7):858–868.
- Luo B, Huang L, Gu Y, et al. Expression of exportin-1 in diffuse large B-cell lymphoma: immunohistochemistry and TCGA analyses. Int J Clin Exp Pathol. 2018;11(12):5547–5560.
- Laín S, Xirodimas D, Lane DP. Accumulating active p53 in the nucleus by inhibition of nuclear export: a novel strategy to promote the p53 tumor suppressor function. Exp Cell Res. 1999 Dec 15;253(2):315–324.
- Kuruvilla J, Savona M, Baz R, et al. Selective inhibition of nuclear export with selinexor in patients with non-Hodgkin lymphoma. Blood. 2017 Jun 15;129(24):3175–3183.
- Chari A, Vogl DT, Gavriatopoulou M, et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N Engl J Med. 2019 Aug 22;381(8):727–738.
- Kalakonda N, Maerevoet M, Cavallo F, et al. Selinexor in patients with relapsed or refractory diffuse large B-cell lymphoma (SADAL): a single-arm, multinational, multicentre, open-label, phase 2 trial. Lancet Haematol. 2020 Jul;7(7):e511–e522.
- Mondello P, Nowakowski GS. Treatment of aggressive B cell lymphomas: updates in 2019. Curr Hematol Malig Rep. 2020 Jun;15(3):225–234.
- Vitolo U, Chiappella A, Franceschetti S, et al. Lenalidomide plus R-CHOP21 in elderly patients with untreated diffuse large B-cell lymphoma: results of the REAL07 open-label, multicentre, phase 2 trial. Lancet Oncol. 2014 Jun;15(7):730–737.
- Vitolo U, Witzig TE, Gascoyne RD, et al. ROBUST: first report of phase III randomized study of lenalidomide/R-CHOP (R2-CHOP) vs placebo/R-CHOP in previously untreated ABC-type diffuse large B-cell lymphoma. Hematol Oncol. 2019;37(S2):36–37.
- Nowakowski GS, Hong F, Scott DW, et al. Addition of lenalidomide to R-CHOP (R2CHOP) improves outcomes in newly diagnosed diffuse large B-cell lymphoma (DLBCL): first report of ECOG-ACRIN1412 a randomized phase 2 US intergroup study of R2CHOP vs R-CHOP. Hematol Oncol. 2019;37(S2):37–38.
- Hagner PR, Man HW, Fontanillo C, et al. CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood. 2015 Aug 6;126(6):779–789.
- Ito T, Ando H, Suzuki T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010 Mar 12;327(5971):1345–1350.
- Chamberlain PP, Lopez-Girona A, Miller K, et al. Structure of the human cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat Struct Mol Biol. 2014 Sep;21(9):803–809.
- Collins I, Wang H, Caldwell JJ, et al. Chemical approaches to targeted protein degradation through modulation of the ubiquitin-proteasome pathway. Biochem J. 2017 Mar 15;474(7):1127–1147.
- Gandhi AK, Kang J, Havens CG, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors ikaros and aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br J Haematol. 2014 Mar;164(6):811–821.
- Lopez-Girona A, Mendy D, Ito T, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012 Nov;26(11):2326–2335.
- Hagner PR, Chiu H, Ortiz M, et al. Activity of lenalidomide in mantle cell lymphoma can be explained by NK cell-mediated cytotoxicity. Br J Haematol. 2017 Nov;179(3):399–409.
- Carpio C, Bouabdallah R, Ysebaert L, et al. Avadomide monotherapy in relapsed/refractory DLBCL: safety, efficacy, and a predictive gene classifier. Blood. 2020 Mar 26;135(13):996–1007.
- Morin RD, Johnson NA, Severson TM, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010 Feb;42(2):181–185.
- Morin RD, Mendez-Lago M, Mungall AJ, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011 Jul 27;476(7360):298–303.
- Béguelin W, Popovic R, Teater M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013 May 13;23(5):677–692.
- Caganova M, Carrisi C, Varano G, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest. 2013 Dec;123(12):5009–5022.
- Sarkozy C, Morschhauser F, Dubois S, et al. A LYSA phase Ib study of tazemetostat (EPZ-6438) plus R-CHOP in patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL) with poor prognosis features. Clin Cancer Res. 2020 Jul 1;26(13):3145–3153.
- Italiano A, Soria JC, Toulmonde M, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 2018 May;19(5):649–659.
- Honma D, Kanno O, Watanabe J, et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 2017 Oct;108(10):2069–2078.
- Prabhu VV, Talekar MK, Lulla AR, et al. Single agent and synergistic combinatorial efficacy of first-in-class small molecule imipridone ONC201 in hematological malignancies. Cell Cycle. 2018;17(4):468–478.
- Sun K, Atoyan R, Borek MA, et al. Dual HDAC and PI3K inhibitor CUDC-907 downregulates MYC and suppresses growth of MYC-dependent Cancers. Mol Cancer Ther. 2017 Feb;16(2):285–299.
- Younes A, Berdeja JG, Patel MR, et al. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: an open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2016 May;17(5):622–631.
- Oki Y, Kelly KR, Flinn I, et al. CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: results from an expanded phase I trial. Haematologica. 2017 Nov;102(11):1923–1930.
- Chan JC, Hannan KM, Riddell K, et al. AKT promotes rRNA synthesis and cooperates with c-MYC to stimulate ribosome biogenesis in cancer. Sci Signal. 2011 Aug 30;4(188):ra56.
- Bywater MJ, Poortinga G, Sanij E, et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012 Jul 10;22(1):51–65.
- Hannan KM, Sanij E, Rothblum LI, et al. Dysregulation of RNA polymerase I transcription during disease. Biochim Biophys Acta. 2013 Mar-Apr;1829(3–4):342–360.
- Poortinga G, Quinn LM, Hannan RD. Targeting RNA polymerase I to treat MYC-driven cancer. Oncogene. 2015 Jan 22;34(4):403–412.
- Khot A, Brajanovski N, Cameron DP, et al. First-in-human RNA polymerase I transcription inhibitor CX-5461 in patients with advanced hematologic cancers: results of a phase i dose-escalation study. Cancer Discov. 2019 Aug;9(8):1036–1049.
- Vose JM, Link BK, Grossbard ML, et al. Phase II study of rituximab in combination with CHOP chemotherapy in patients with previously untreated, aggressive non-Hodgkin’s lymphoma. J Clin Oncol. 2001 Jan 15;19(2):389–397.
- Coiffier B, Lepage E, Briere J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002 Jan 24;346(4):235–242.
- Habermann TM, Weller EA, Morrison VA, et al. Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. J Clin Oncol. 2006 Jul 1;24(19):3121–3127.
- Pfreundschuh M, Trümper L, Osterborg A, et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera international trial (MInT) group. Lancet Oncol. 2006 May;7(5):379–391.
- van Imhoff GW, McMillan A, Matasar MJ, et al. Ofatumumab versus rituximab salvage chemoimmunotherapy in relapsed or refractory diffuse large B-cell lymphoma: the ORCHARRD study. J Clin Oncol. 2017 Feb 10;35(5):544–551.
- Gisselbrecht C, Glass B, Mounier N, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010 Sep 20;28(27):4184–4190.
- Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017 Oct 19;130(16):1800–1808.
- Jurczak W, Zinzani PL, Gaidano G, et al. Phase IIa study of the CD19 antibody MOR208 in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma. Ann Oncol. 2018 May 1;29(5):1266–1272.
- Salles G, Duell J, González Barca E, et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020 Jul;21(7):978–988.
- Choi Y, Diefenbach CS. Polatuzumab vedotin: a new target for B cell malignancies. Curr Hematol Malig Rep. 2020 Apr;15(2):125–129.
- Sehn LH, Herrera AF, Flowers CR, et al. Polatuzumab vedotin in relapsed or refractory diffuse large B-cell lymphoma. J clin oncol. 2020;38(2):155–165.
- Zammarchi F, Corbett S, Adams L, et al. ADCT-402, a PBD dimer-containing antibody drug conjugate targeting CD19-expressing malignancies. Blood. 2018 Mar 8;131(10):1094–1105.
- Kahl BS, Hamadani M, Radford J, et al. A phase i study of ADCT-402 (loncastuximab tesirine), a novel pyrrolobenzodiazepine-based antibody-drug conjugate, in relapsed/refractory B-cell non-hodgkin lymphoma. Clin Cancer Res. 2019 Dec 1;25(23):6986–6994.
- Trnĕný M, Verhoef G, Dyer MJ, et al. A phase II multicenter study of the anti-CD19 antibody drug conjugate coltuximab ravtansine (SAR3419) in patients with relapsed or refractory diffuse large B-cell lymphoma previously treated with rituximab-based immunotherapy. Haematologica. 2018 Aug;103(8):1351–1358.
- Coiffier B, Thieblemont C, de Guibert S, et al. A phase II, single-arm, multicentre study of coltuximab ravtansine (SAR3419) and rituximab in patients with relapsed or refractory diffuse large B-cell lymphoma. Br J Haematol. 2016 Jun;173(5):722–730.
- Advani RH, Lebovic D, Chen A, et al. Phase I study of the anti-CD22 antibody-drug conjugate pinatuzumab vedotin with/without rituximab in patients with relapsed/refractory B-cell non-Hodgkin lymphoma. Clin Cancer Res. 2017 Mar 1;23(5):1167–1176.
- Advani R, Chen AI, Lebovic D, et al. Final results of a phase i study of the anti-CD22 antibody-drug conjugate (ADC) DCDT2980S with or without rituximab (RTX) in patients (Pts) with relapsed or refractory (R/R) B-cell non-Hodgkin’s lymphoma (NHL). Blood. 2013;122(21):4399.
- Morschhauser F, Flinn IW, Advani R, et al. Polatuzumab vedotin or pinatuzumab vedotin plus rituximab in patients with relapsed or refractory non-Hodgkin lymphoma: final results from a phase 2 randomised study (ROMULUS). Lancet Haematol. 2019 May;6(5):e254–e265.
- Fabbri M, Smart C, Pardi R. T lymphocytes. Int J Biochem Cell Biol. 2003 Jul;35(7):1004–1008.
- Labrijn AF, Janmaat ML, Reichert JM, et al. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019 Aug;18(8):585–608.
- Nagorsen D, Kufer P, Baeuerle PA, et al. Blinatumomab: a historical perspective. Pharmacol Ther. 2012 Dec;136(3):334–342.
- Haas C, Krinner E, Brischwein K, et al. Mode of cytotoxic action of T cell-engaging BiTE antibody MT110. Immunobiology. 2009;214(6):441–453.
- Goebeler ME, Knop S, Viardot A, et al. Bispecific T-cell engager (BiTE) antibody construct blinatumomab for the treatment of patients with relapsed/refractory non-hodgkin lymphoma: final results from a phase i study. J Clin Oncol. 2016 Apr 1;34(10):1104–1111.
- Viardot A, Goebeler ME, Hess G, et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood. 2016 Mar 17;127(11):1410–1416.
- Bacac M, Fauti T, Sam J, et al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin Cancer Res. 2016 Jul 1;22(13):3286–3297.
- Hutchings M, Iacoboni G, Morschhauser F, et al. CD20-Tcb (RG6026), a novel “2:1” format T-cell-engaging bispecific antibody, induces complete remissions in relapsed/refractory B-cell non-Hodgkin’s lymphoma: preliminary results from a phase i first in human trial. Blood. 2018;132(Supplement 1):226.
- Dickinson MJMF, Iacoboni G, Carlo-Stella C, et al. CD20-TCB in relapsed or refractory non-Hodgkin lymphoma: durable complete responses and manageable safety observed at clinically relevant doses in phase I dose escalation hemasphere. 2020;4:S1:80.
- Hutchings M, Gritti G, Sureda A, et al. CD20-TCB, a novel T-cell-engaging bispecific antibody, can be safely combined with the anti-PD-L1 antibody atezolizumab in relapsed or refractory B-cell non-Hodgkin lymphoma. Blood. 2019;134(Supplement_1):2871.
- Morschhauser F, Carlo-Stella C, Offner F, et al. Dual CD20-targeted therapy with concurrent CD20-TCB and obinutuzumab shows highly promising clinical activity and manageable safety in relapsed or refractory B-cell non-hodgkin lymphoma: preliminary results from a phase ib trial. Blood. 2019;134(Supplement_1):1584.
- Smith EJ, Olson K, Haber LJ, et al. A novel, native-format bispecific antibody triggering T-cell killing of B-cells is robustly active in mouse tumor models and cynomolgus monkeys. Sci Rep. 2015 Dec 11;5:17943.
- Bannerji R, Brown JR, Advani RH, et al. Phase 1 study of REGN1979, an anti-CD20 x anti-CD3 bispecific monoclonal antibody, in patients with CD20+ B-cell malignancies previously treated with CD20-directed antibody therapy. Blood. 2016;128(22):621.
- Schuster SJ, Bartlett NL, Assouline S, et al. Mosunetuzumab induces complete remissions in poor prognosis non-hodgkin lymphoma patients, including those who are resistant to or relapsing after chimeric antigen receptor T-cell (CAR-T) therapies, and is active in treatment through multiple lines. Blood. 2019;134(Supplement_1):6.
- Engelberts PJ, Hiemstra IH, de Jong B, et al. DuoBody-CD3xCD20 induces potent T-cell-mediated killing of malignant B cells in preclinical models and provides opportunities for subcutaneous dosing. EBioMedicine. 2020;52:102625.
- Patel K, Michot J-M, Chanan-Khan AA, et al. Preliminary safety and anti-tumor activity of XmAb13676, an anti-CD20 x anti-CD3 bispecific antibody, in patients with relapsed/refractory non-hodgkin’s lymphoma and chronic lymphocytic leukemia. Blood. 2019;134(Supplement_1):4079.
- Mohty M, Dulery R, Gauthier J, et al. CAR T-cell therapy for the management of refractory/relapsed high-grade B-cell lymphoma: a practical overview. Bone Marrow Transplant. 2020 Aug;55(8):1525–1532.
- Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019 Jan 3;380(1):45–56.
- Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017 Dec 28;377(26):2531–2544.
- Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019 Jan;20(1):31–42.
- Abramson JSGL, Palomba ML, Lunning MA, et al. Updated safety and long term clinical outcomes in TRANSCEND NHL 001, pivotal trial of lisocabtagene maraleucel (JCAR017) in R/R aggressive NHL. J clin oncol. 2018;36(15_suppl):7505.
- Abramson JS, Palomba ML, Gordon LI, et al. Pivotal safety and efficacy results from transcend NHL 001, a multicenter phase 1 study of lisocabtagene maraleucel (liso-cel) in relapsed/refractory (R/R) large B cell lymphomas. Blood. 2019;134(Supplement_1):241.
- Ruella M, Maus MV. Catch me if you can: leukemia escape after CD19-directed T cell immunotherapies. Comput Struct Biotechnol J. 2016;14:357–362.
- Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018 Oct;8(10):1219–1226.
- Zah E, Lin MY, Silva-Benedict A, et al. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. 2016 Jun;4(6):498–508.
- Ruella M, Barrett DM, Kenderian SS, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest. 2016 Oct 3;126(10):3814–3826.
- Schneider D, Xiong Y, Wu D, et al. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J Immunother Cancer. 2017;5:42.
- Shah NN, Zhu F, Taylor C, et al. A phase 1 study with point-of-care manufacturing of dual targeted, tandem anti-CD19, anti-CD20 chimeric antigen receptor modified T (CAR-T) cells for relapsed, refractory, non-hodgkin lymphoma. Blood. 2018;132(Supplement 1):4193.
- Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018 Jan;24(1):20–28.
- Hossain N, Sahaf B, Abramian M, et al. Phase I experience with a bi-specific CAR targeting CD19 and CD22 in adults with B-cell malignancies. Blood. 2018;132(Supplement 1):490.
- Ardeshna K, Marzolini MAV, Osborne W, et al. Study of AUTO3, the first bicistronic chimeric antigen receptor (CAR) targeting CD19 and CD22, followed by anti-PD1 consolidation in patients with relapsed/refractory (r/r) diffuse large B cell lymphoma (DLBCL): Alexander study. Blood. 2018;132(Supplement 1):1679.
- Zhao Z, Condomines M, van der Stegen SJC, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015 Oct 12;28(4):415–428.
- Park JH, Palomba ML, Batlevi CL, et al. A phase i first-in-human clinical trial of CD19-targeted 19-28z/4-1BBL “armored” CAR T cells in patients with relapsed or refractory NHL and CLL including richter’s transformation. Blood. 2018;132(Supplement 1):224.
- Santomasso BD, Park JH, Salloum D, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. 2018 Aug;8(8):958–971.
- Xu Y, Yang Z, Horan LH, et al. A novel antibody-TCR (AbTCR) platform combines Fab-based antigen recognition with gamma/delta-TCR signaling to facilitate T-cell cytotoxicity with low cytokine release. Cell Discov. 2018;4:62.
- Liu H, Horan LH, Grupp SA, et al. Anti-CD19 ARTEMIS™ T cells prevent excessive inflammatory cytokine release, including IL-6, in a co-culture model of CRS. Cancer Res. 2018;78(13 Supplement):1537.
- Ying Z, Long L, Liu H, et al. ET190L1-ARTEMIS T cell therapy to induce complete remission of relapsed and refractory (r/r) B-cell lymphoma with no cytokine release syndrome in the first-in-human clinical study. J clin oncol. 2018;36(15_suppl):3049.
- Ying Z, Long L, Liu H, et al. ET190L1-artemistm T cell therapy results in durable disease remissions with no cytokine release syndrome or neurotoxicity in patients with relapsed and refractory B-cell lymphoma. Blood. 2018;132(Supplement 1):1689.
- Ruggeri L, Capanni M, Casucci M, et al. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood. 1999 Jul 1;94(1):333–339.
- Ruggeri L, Capanni M, Urbani E, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002 Mar 15;295(5562):2097–2100.
- Asai O, Longo DL, Tian ZG, et al. Suppression of graft-versus-host disease and amplification of graft-versus-tumor effects by activated natural killer cells after allogeneic bone marrow transplantation. J Clin Invest. 1998 May 1;101(9):1835–1842.
- Szmania S, Lapteva N, Garg T, et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother. 2015 Jan;38(1):24–36.
- Shah N, Li L, McCarty J, et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol. 2017 May;177(3):457–466.
- Liu E, Marin D, Banerjee P, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020 Feb 6;382(6):545–553.
- Moia R, Patriarca A, Schipani M, et al. Precision medicine management of chronic lymphocytic leukemia. Cancers (Basel). 2020 Mar 10;12(3):642.
- Moia R, Patriarca A, Deambrogi C, et al. An update on: molecular genetics of high-risk chronic lymphocytic leukemia. Expert Rev Hematol. 2020 Feb;13(2):109–116.
- Rossi D, Diop F, Spaccarotella E, et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood. 2017 Apr 6;129(14):1947–1957.
- Rossi D, Spina V, Bruscaggin A, et al. Liquid biopsy in lymphoma. Haematologica. 2019 Apr;104(4):648–652.
- Kurtz DM, Esfahani MS, Scherer F, et al. Dynamic risk profiling using serial tumor biomarkers for personalized outcome prediction. Cell. 2019 Jul 25;178(3):699–713 e19.
- Kurtz DM, Scherer F, Jin MC, et al. Circulating tumor DNA measurements as early outcome predictors in diffuse large B-cell lymphoma. J Clin Oncol. 2018 Oct 1;36(28):2845–2853.
- Nastoupil LJ, Jain MD, Feng L, et al. Standard-of-care axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma: results from the US lymphoma CAR T consortium. J Clin Oncol. 2020 Sep 20;38(27):3119-3128.